|Articles|January 1, 2016
- LCGC North America-01-01-2016
- Volume 34
- Issue 1
Glycosylation in mAb Therapeutic Products: Analytical Characterization and Impact of Process

Glycosylation of monoclonal antibody (mAb) therapeutics is widely recognized by the regulators and the industry as a critical quality attribute (CQA). Hence, it is necessary that glycosylation is measured and adequately controlled during production. This installment reviews the various process parameters and raw material attributes that affect glycosylation, as well as the different analytical tools that are used for characterization, with greater emphasis on the chromatographic methods of analysis. Key recent advancements that have occurred in the past five years are also discussed briefly. While significant progress has been made in the monitoring of glycosylation, its real time control has yet to be demonstrated.
Advertisement
Glycosylation of monoclonal antibody (mAb) therapeutics is widely recognized by the regulators and the industry as a critical quality attribute (CQA). Hence, it is necessary that glycosylation is measured and adequately controlled during production. This installment reviews the various process parameters and raw material attributes that affect glycosylation, as well as the different analytical tools that are used for characterization, with greater emphasis on the chromatographic methods of analysis. Key recent advancements that have occurred in the past five years are also discussed briefly. While significant progress has been made in the monitoring of glycosylation, its real time control has yet to be demonstrated.
Biotherapeutics or biologics have emerged as potent treatments of a variety of diseases, including cancer and other immunological disorders. Since these products are produced in biological systems, they often exhibit a high degree of variability (physical and chemical modifications). These modifications are known to potentially impact the characteristics and biological activity of the therapeutic drug. Additionally, attributes such as drug clearance and half-life are also affected by these modifications. Regulatory authorities mandate manufacturers to make efforts to minimize these heterogeneities during process and formulation development, so as to ensure that the safety of patients is not jeopardized and that the drug efficacy stays intact. To this effect, the concept of quality by design (QbD) encourages designing quality into the product (1,2). Under this approach, any physical and chemical changes in the drug product that have the potential to affect the safety and efficacy are considered critical quality attributes (CQAs). And, manufacturing is accordingly designed so that these CQAs are consistently monitored and controlled (3–5).
Among the different CQAs (generally only a few for microbial products and more for mammalian products), a glycosylation profile of a product is well-recognized as a CQA that requires stringent monitoring and control. Glycosylation refers to a sequence of events that take place in the Golgi body and the endoplasmic reticulum of the cells of the expression system (predominantly mammalian) that lead to post-translational addition and processing of carbohydrate or glycan moieties to the protein backbone (usually serine- or threonine-linked glycosylation for O-linked glycans and asparagine-linked glycosylation for N-linked glycans) (6). The addition of a precursor 13-residue oligosaccharide moiety GlcNAc2Man8Gluc3 to the nascent polypeptide chain occurs in the endoplasmic reticulum, following which the assembly folds and acquires its secondary, tertiary, and quaternary structure. The folded structure gets transported via vesicles to the Golgi bodies (7). A cooperative and competitive interaction between multiple enzymes, cell lines, and cell culture conditions causes processing of the oligosaccharide structure during which some of the already attached moieties like mannose are trimmed to yield a five-residue core (Man3GlcNAc2), to which N-acetylglucosamine (GlcNAc), fucose, galactose, and N-acetylneuraminic acid (sialic acid or Neu5Ac) residues get added in a probabilistic fashion.
These moieties are critical in terms of their role in in-vivo safety and efficacy (pharmacodynamics and pharmacokinetics), protein folding, protein targeting and trafficking, ligand binding, stability, protein half-life regulation, and immunogenicity (8). For instance, researchers have shown that the fucosylation of monoclonal antibodies (mAbs) causes reduced antibody-dependent cell mediated cytotoxicity (ADCC). Similarly, some of the therapeutic mAbs with non-galactosylated glycan profiles have shown less affinity toward the c1q component of the complement cascade, thus reducing the complement dependent cytotoxicity (CDC). A good number of mAb therapeutics that are used for treating autoimmune disorders have exhibited a strong and positive correlation between anti-inflammatory effects and the presence of the terminal sialic acid residues on the oligosaccharide structure attached to the protein polypeptide backbone (9). Thus, depending on the mechanism of action (MoA), CQA is defined for the therapeutic-that is, if ADCC is a desired therapeutic effect, fucosylation is selected as the CQA. Similarly, for CDC and immunomodulation, galactosylation and sialylation, respectively, are the CQAs (10).
Given the criticality of their role in exertion of a therapeutic protein’s pharmacological effect, it is necessary to characterize detailed glycan structures, as well as those specific glycan subsets that are known or are likely to cause adverse events in the body. Notwithstanding the significant advancements in analytical characterization of these products, glycosylation analysis continues to be nontrivial and demands considerable resources in terms of analyst expertise as well as high-end characterization tools. In this installment, we review the various analytical tools that are used for characterization of mAb glycosylation, with a greater emphasis on the chromatographic methods of analysis. Key recent advancements that have occurred in the past five years are also discussed briefly.
Regulatory Perspective
Detailed workflow and regulatory expectations have already been laid out in the various regulatory guidance documents governing production, commercialization, and licensure of biologic drugs (11–14). The International Conference on Harmonization (ICH) guidelines mandate characterization of total carbohydrate contents (including neutral contents, amino sugars, and sialic acid), antennary profile, glycosylation sites interglycosidic linkages, and anomeric configuration (11). European Medical Agency (EMA) guidelines have an additional requirement of presenting data with regards to the presence or absence of glycosylation in light chain, degree of mannosylation, galactosylation, fucosylation, and sialylation (12). In the event that a product is found to contain any glycosylated variant that differs from the reference product, it is essential to establish that such differences are noncritical with respect to the safety and efficacy of the therapeutic using more comprehensive preclinical and clinical data. Figure 1 illustrates the framework for assessing the change in the glycan profiles of the biotherapeutic in question.
Figure 1: Framework for assessing changes in the glycan profiles for mAb therapeutics.
The use of orthogonal, high resolution, and innovative analytical tools that allow us to decipher product heterogeneity, as well as consistency at batch-to-batch levels is mandatory. Computational biology (bioinformatics) has further emerged as a source of information for assessing immunogenicity potential of a biotherapeutic (15). These activities allow for a rigorous and comprehensive review of the product development data by the regulatory bodies, based on scientific merit, and renders decisions that are data driven.
Process Parameters that Affect Glycosylation
Glycosylation is known to be affected by numerous process parameters, as well as genetic elements (16,17). However, in this column installment we focus our discussion only on important process parameters that either impact yield or the overall quality of the product (18–38).
A summary of important process variables affecting the glycosylation is presented in Table I. Based on literature review, it is evident that out of the various factors that affect glycosylation, the concentration of manganese, temperature, and concentration of ammonium chloride are recognized to have a major impact on glycosylation. Manganese supplementation under low glucose feed have resulted in higher levels of galactosylated and mannosylated glycoforms on mAbs expressed in Chinese hamster ovary (CHO) cells. Mn2+ is known to act as a cofactor to β-1,4-galactosyltransferase, which selectively up-regulates galactosylation of protein therapeutics. However, when glucose feed is substituted with other sugars such as galactose and fructose, specifically higher mannosylated species (M5 mannose glycans) have been observed, because of limited availability of nucleotide sugars. Fucosylated glycoforms remain consistent regardless of the manganese concentration in the culture media (23–26).
With regard to the effect of temperature, it is generally observed that a downshift in temperature helps in lowering the content of acidic charge variant content in the mAb therapeutics (27). A recent study showed that good correlation exists between charge and glycosylation profiles of mAbs (28). However, an elaborate description of the mechanism of how such an affect manifests is not well-understood. Transcriptomic studies, to a large extent, provide certain insights into such an observation. It is shown that the downshift of temperature affects the gene expressions of enzyme systems (glycosyl transferases, mannosidase, and galactosyltransferase), which affects their accessibility to cause the necessary terminal site modification in mAbs (such as N–terminal pyroglutamate formation and C–terminal lysine clipping) and thus explains the varying glycosylation profiles observed among these variants (29,30,39). Apart from this, a negative correlation has been found to exist between terminal glycoforms (sialic acid) and DO levels in the culture (31,32).
Ammonium chloride is also recognized as a key component that is shown to play a critical role in the addition of terminal sugars (sialylation) during the glycosylation process (35–37). An ammonia concentration typically below 2 mM increases the intracellular pH, which in turn leads to an inhibition of galactosyl transferase and sialyl transferase, enzymes that mediate addition of the terminal sugars (38,40).
While pH has long been believed to be a critical process parameter during cell culture, the optimum pH range for cell culture is believed to be 6.9–8.2, with any perturbance beyond this range affecting glycosylation. Lower pH has been known to result in more galactosylation and hence production of a higher level of the corresponding species (33,34).
Various sugars, such as glucose, mannose, and galactose, have been known to affect mannosylation, fucosylation, and sialylation. It has been reported that addition of galactose to the culture medium improves sialylation. This improvement results from an increased availability of the binding sites (galactose moieties) for sialic residues (20). The ratio of mannose to glucose has also been known to affect mannosylation. A higher ratio (0.94) has been shown to result in increased levels of mannosylated species (18).
Characterization of mAb Glycosylation
Characterization of glycoproteins and released glycans involves the use of a large number of high resolution, orthogonal analytical tools, that together provide a complete glycan fingerprint at three levels. The first level identifies possible glycosylation sites (macro heterogeneity) present on the mAb (Cγ2 domains of Fc fragment, VL domain and VH domain or both). Reduction and acidification, papain digestion, or chemical digestion are usually performed to determine the oligosaccharide structures present on each peptide fragment (41,42).
The second level of information is with regards to the variation in the sequence of the sugars (microheterogeneity). This information is obtained either by cleaving the protein backbone and analyzing the glycopeptides or by enzymatically releasing the carbohydrate moieties from the protein backbone (7). The first approach is accomplished by use of proteolytic enzymes like trypsin, lysyl endopeptidase (Lys-C), endoproteinase Glu-C, and pronase. In the second approach, enzymes such as PNGase F (releases all N-linked oligosaccharides), PNGase A (releases glycan with α1-3 core-fucosylation), and endoglycosidase H (releases high mannose like N-glycans) are used (10). Apart from these, two O-glycanases are also commercially available for cleaving the Galβ1-3GalNAc linked to serine or threonine residues, but the presence of sialic acid, fucose, GlcNAc, and GalNAc residues prevent this cleavage. The use of an enzymatic method is limited because of the variable structure of the O-glycans and the narrow substrate specificity of the available enzymes (43).
Both O- and N-glycans can be released by chemical hydrazinolysis. However, the drawback associated with this method is the degradation of reducing end monosaccharides (β-elimination) and destruction of noncarbohydrate substituents. Moreover, alditols obtained by β-elimination also prevent subsequent labeling of the oligosaccharide that hinders detection of glycans during separation and fractionation (44). The released glycans are recovered from the pool of peptides, salts, and other compounds by various methods based on the differences in the polarity and functionality of peptides and glycans. These methods include ethanol precipitation, zwitterionic hydrophilic interaction liquid chromatography (HILIC), solid-phase extraction (SPE) using graphitized carbon black (GCB), titanium, or anion-exchange chromatography using borate resin. This is followed by a step involving specific entrapment of the separated glycans. Furthermore, the separated glycans are then derivatized using one of several possible approaches, with the most popular being reductive amination with several types of aromatic amines. In this approach, the reducing-end of the oligosaccharides is labeled with either a fluorescence tag or a UV-absorbing tag for further detection and quantification. The labeling agent used generally depends on the reaction condition (aqueous or nonaqueous) and the analytical method used for further separation. Possible agents include 2-aminopyridine (PA), 2-aminobenzamide (2-AB), 2-aminobenzoic acid (2-AA), 8-aminopyrene-1,3,6-trisulfonate (APTS), and 1,2-diamino-4,5-methylene-dioxybenzene-dihydrochloride (DMB) (45). An alternative approach is permethylation of glycans to prevent loss of sialic acid during mass spectrometry (MS)-based analysis (45).
Reductively aminated derivatives are generally chemically stable and most of them, except for 2-aminopyridine and 8-aminonaphthalene-1,3,6-trisulfonate (ANTS), can be converted back to free oligosaccharides by treatment with hydrogen peroxide or acetic acid (46). Finally, the released, derivatized or un-derivatized glycans can be separated using a chromatographic technique (discussed in subsequent sections), such that the resolution and retention depend on the affinity of the glycan for the matrix used for separation followed by fluorescence or MS detection. It should be noted that analysis of native, unlabeled, and reduced (un-derivatized) glycans can also be performed but this analysis is restricted because of the limited resolution that the existing separation tools offer.
The third level of glycan structural analysis involves unraveling information regarding the intermolecular structures of oligosaccharides by characterization of the linkages between the constituting monosaccharides of glycans. A sequential enzyme-catalyzed reaction is performed on the basis of the bonds that occur between different monosaccharides, and the products are quantified using various analytical techniques to analyze the bond structure of the original glycan (7). The various chromatographic techniques used for the separation of released, purified, and labeled or unlabeled glycans are described below, and a workflow for glycan identification and characterization is illustrated in Figure 2.
Figure 2: Workflow of glycan profiling. Glycans from various sources, such as proteins or mAbs are first released by chemical or enzymatic methods. This is then followed by isolation of the released glycans, and sample clean-up for removal of salts or denaturants. Labeling with appropriate reagents is performed so as to improve detectability. Various chromatographic tools, as shown in the figure, are used for separation of the glycans. Detection is generally done using a fluorescence detector or high-resolution MS.
Separation-Based Analysis of mAb Glycans
Like any other separation, oligosaccharide separation is principally done based on the properties of sample components such as size, polarity, and ionic strength. Predominantly, there are four separation-based techniques that are used for the characterization of mAb glycoproteins: reversed-phase high performance liquid chromatography (HPLC), HILIC, anion-exchange chromatography, and high performance anion-exchange chromatography.
Reversed-Phase HPLC
Reversed-phase HPLC refers to a mode of separation using nonpolar stationary phases and polar mobile phases. The polar nature of oligosaccharides causes minimal retention on reversed-phase columns resulting in their simultaneous elution and thus hampering the separation. To overcome this, glycans are derivatized into more hydrophobic forms by introducing a chromophore through the hydroxyl group (generally PA, 2-AB, or 2-AA) or by permethylation (7,47). However, labeling oligosaccharides with bulky hydrophobic tags has not been particularly successful, because it impairs separation based on structural differences among sugar moieties and causes the elution of oligosaccharides in the order of decreasing size. However, PA is still regarded as one of the most efficient labeling reagents because of its low hydrophobicity and minimal effect on retention time (48). Derivatization by permethylation has also emerged as a viable option for reversed-phase HPLC separations of sialylated glycans because of the increased stability, enhanced ionization efficiency, and predictable fragmentation when coupled to a mass spectrometer (49). However, a rigorous optimization of this step is necessary because it is prone to form various reaction by-products and impurities that may hinder the separation process (50).
The biggest advantage of reversed-phase HPLC is its compatibility for direct hyphenation to MS. This is because of the fact that the buffers that are generally used in reversed-phase separations allow for simultaneous separation during chromatography and ionization of the analyte during the MS. A major limitation of glycan separation using reversed-phase HPLC, however, is its limited ability to resolve glycan structural isomers. An alternative method is that of using porous graphitic carbon (PGC) as the stationary phase for chromatographic separation of labelled glycans,oligosaccharide alditols, and even permethylated glycans (44,51,52). This is a method of choice for direct liquid chromatography–mass spectrometry (LC–MS) of O-glycans released by β-elimination and is efficient in separating structural isomers (53). Further, for better separation and structural assignment, reversed-phase HPLC is often used together with HILIC and anion-exchange chromatography as a multidimensional strategy (44,51). Hydrophobic interaction chromatography (HIC) has also been used for the separation of glycoproteins on the basis of glycoforms (54).
HILIC
HILIC, in contrast to reversed-phase HPLC, uses a relatively polar mobile phase and a polar stationary phase (for example, silica derivatized with amide–amine groups). The polar nature of oligosaccharides enables them to interact with the polar stationary phase based on dipole–dipole interaction, electrostatic interactions, and hydrogen bonding. Therefore, HILIC separates sugar moieties as a function of their capacity to form hydrogen bonds with an amine–amide bonded-silica phase (7,43). Hydrophobic peptides are washed off with the organic mobile phase, while polar glycopeptides are retained on the HILIC phase, and these are eluted by increasing the water content of the mobile phase, thus separating glycosylated and nonglycosylated peptides (55).
Unlike reversed-phase HPLC, the purpose of labeling in HILIC is primarily for enhancing the sensitivity of detection. Because of the correlation between the glycan size (based on the addition of monosaccharide units to oligosaccharides) and its retention time, it is sometimes referred to as size-based separation (55). Oligosaccharides labeled with PA, 2-AB, 2-AA, and acridine derivatives are easily compatible with HILIC conditions, though with different retention times for derivatized and unmodified sugar structures (47). Some glycan structural isomers (G1Fa and G1Fb, differing in position of galactose residue) have been successfully resolved using HILIC (56). The structural variation of the individual glycans and changes in retention times can possibly be predicted by comparing the elution time of eluted glycans with a labeled glucose ladder (partial dextran hydrolysate or a mixture of oligosaccharides) (51).
HILIC is compatible with both fluorescence and MS detection methods (electrospray ionization [ESI] or matrix assisted laser desorption-ionization [MALDI]-time of flight [TOF]-TOF-MS-MS), where when combined with fluorescence detection of 2-AB labeled glycans, it is considered to be a gold standard for analyzing N-glycans of biotherapeutics (56). When coupled to MS, no tagging of glycans is needed, thereby yielding a much simpler procedure (47). For in-depth analysis of complex oligosaccharide mixtures, a multidimensional HPLC approach can be applied by combining HILIC with other chromatographic methods, such as anion exchange or weak anion exchange, that give comprehensive separation of complex N-glycans (57).
Anion Exchange and High-Performance Anion Exchange
Because carbohydrates are weak acids (pKa > 11), they can be retained on a strong anion exchange column (pH > 13), and hence can be separated using high performance (high pH) anion-exchange chromatography. With this tool, glycan moieties are differentially adsorbed on a positively charged matrix, influenced by the number of acidic groups, molecular size, sugar composition, and monosaccharide linkage (43). Elution is often done by a salt gradient-that is, using concentration gradients of a sodium hydroxide–sodium acetate system. Because of the use of a high concentration of salt, the application of this system to MS is restricted (51). Detection is generally accomplished by pulsed amperometric detection (PAD), which is based on measuring the current generated via oxidation or reduction reactions on the electrode surface. The detection is highly sensitive, and concentrations of oligosaccharides can be measured by comparing its response with a calibration curve of reference compounds. However, only relative quantitation is possible because of the lack of reference compounds for generating the calibration curve (7).
Anion-exchange or weak anion-exchange HPLC can also separate some labeled (2-AB or PA) glycans, and these are generally performed from neutral to slightly alkaline pH. Separation is based on the negative charge of glycans, which in turn is determined by the number of sialic acids, carboxylic acids, glucoronic acids, sulfates, and phosphates (51). Elution is achieved via a salt gradient followed by fluorescence-based detection. Complex glycans on erythropoietin are generally characterized using this technique (58). Weak anion-exchange HPLC and high performance anion-exchange chromatography can also be combined to achieve a multidimensional separation (57).
Monitoring and Control of Glycosylation
As is evident from the aforementioned considerations, monitoring and control of glycosylation throughout the life cycle of a product, from cell line development to final product manufacturing, is of paramount importance (59,60). During cell line screening and early upstream process development, glycosylation analysis is required to select the best probable clone, as well as optimized culture conditions, from which a properly glycosylated product is obtained. Although there are very few reports on real time analysis of glycosylation (24,26,61), efforts are ongoing toward devising means or methods for achieving this. In view of the factors affecting glycosylation, monitoring can be performed either at the gene level, using commercially available gene chips, or at the protein level using the various techniques that have been described in detail in the previous sections. A major hurdle is the significant time that is spent on sample preparation. Over time, researchers have succeeded in shortening the analysis time and making the analysis automatic, using robotic sampling systems that offer high throughput glycan analysis and brings us closer to achieving real time monitoring and control of glycosylation (62–65).
A comparison of transcriptome and proteome of expressed genes (using gene chips, cDNA microarrays, real-time polymerase chain reaction [PCR] and two-dimensional [2D] gel electrophoresis–MS approaches), has led to the conclusion that the metabolic shift of the cells during culture is the combinatorial effect of biochemical reactions, as well as transcriptional events (61). Further, it has been observed that galactosylation of mAb N-glycans in CHO cell lines can be predicted accurately enough, and it can be controlled by varying supplements (such as uridine and manganese chloride) and cell culture conditions, using statistical modeling and measurement of cell surface glycans, so as to predict product glycosylation (26). These types of integrated, knowledge-based approaches are necessary and enable deeper understanding, monitoring, and control of glycosylation events.
Although most researchers have taken into consideration the mesoscale (within the cytoplasm) and macroscale (within the reactor) variables to study their effect on glycosylation patterns, it is of prime importance to look at intracellular variables that affect glycosylation. As far as modeling is considered, a detailed understanding of all the micro-, meso-, and macroscale variables is a necessary prerequisite. An assessment of controllability of glycosylation, by modeling the effect of various intracellular variables such as enzymes, nucleotides, and sugar donors on the final pattern of glycosylation, has recently been published (66). The use of manganese chloride, ammonium chloride, and galactose as mesoscale input variables has shown that glycosylation control is possible during cell culture. Multivariate data analysis with parallel factor analysis (PARAFAC), principal component analysis (PCA), multiway PCA, partial least squares (PLS), and multiway PLS has been successfully used to optimize cell culture conditions (67,68). These models assist not only in cell line development but also enhance the speed and accuracy, which enables optimization of the media and cell culture conditions in a reproducible and consistent manner.
As already discussed above, understanding the dynamics of glycosylation requires real time glycosylation analysis. To this end, a new platform method has been described recently for real-time glycan analysis that comprises a sampling probe and an ultrahigh-pressure liquid chromatography system (µSI-Glycan map-UPLC, Waters Corporation) that allows automated sampling and sample preparation for glycan analysis in real time (24). Data can be collected every 12 h versus 1 day in the case of off-line analyzers for intact glycans, with varying degrees of mannosylation, fucosylation, and galactosylation.
Conclusions
Glycosylation is an important CQA, and hence it is necessary to monitor and control it throughout the product life cycle. Heterogeneity in glycosylation can be minimized by carefully controlling the various critical parameters like ammonium chloride, manganese, temperature, pH, and various sugars during cell culture. Orthogonal, analytical techniques must be used to analyze the heterogeneity at various levels of glycosylation. Current research focuses mainly on automating the process of monitoring and control of glycosylation in cell culture. In the future, this research may lead to a holistic approach to control this CQA, which will greatly help us in achieving consistency in product quality. While significant progress has been made in monitoring of glycosylation, its real-time control has yet to be demonstrated.
References
- A.S. Rathore and H. Winkle, Nat. Biotechnol.27(1), 26–34 (2009).
- A.S. Rathore, Trends in Biotechnol. 27(9), 546–553 (2009).
- E.K. Read, J.T. Park, R.B. Shah, B.S. Riley, K.A. Brorson, and A.S. Rathore, Biotechnol. Bioen.105(2) 276–284 (2010).
- E.K. Read, J.T. Park, R.B. Shah, B.S. Riley, K.A. Brorson, and A.S. Rathore, Biotechnol. Bioeng.105(2), 285–295 (2010).
- A.S. Rathore, R. Bhambure, and V. Ghare, Anal. Bioanal. Chem.398(1), 137–154 (2010).
- P. Hossler, F.K. Sarwat, and L.J. Zheng, Glycobiology19(9), 936–949 (2009).
- I.J. Del Val, C. Kontoravdi, and J.M. Nagy, Biotechnol. Progr.26(6), 1505–1527 (2010).
- G. Walsh and J. Roy, Nat. Biotechnol.24(10), 1241–1252 (2006).
- A. EonâDuval, H. Broly, and R. Gleixner, Biotechnol. Progr.28(3), 608–622 (2012).
- C. Huhn, M.H. Selman, L.R. Ruhaak, A.M. Deelder, and M. Wuhrer, Proteomics9(4), 882–913 (2009).
- International Conference on Harmonization, ICH Q6B Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products, EMA Document CPMP/ICH/365/96 (ICH, Geneva, Switzerland, 1999).
- European Medicine Agency, Guideline on Development, Production, Characterization and Specifications for Monoclonal Antibodies and Related Products, EMEA/CHMP/BWP/157653/2007 (EMA, London, England, 2007).
- US Food and Drug Administration, Guidance for Industry, Q8 Pharmaceutical Development (FDA, Rockville, Maryland, 2006).
- US Food and Drug Administration, Guidance for Industry, Q9 Quality Risk Management (FDA, Rockville, Maryland, 2006).
- C. BaileyâKellogg, A.H. Gutiérrez, L. Moise, F. Terry, W.D. Martin, and A.S. De Groot, Biotechnol. Bioeng. 111(11), 2170–2182 (2014).
- S.B. Richardson, Genet. Eng. Biotechnol. News35, 28–29 (2015).
- M. Spearman and M. Butler in Animal Cell Culture, M. Al-Rubeai, Ed. (Springer International Publishing, 2015), pp. 237–258.
- C.J. Huang, H. Lin, and J.X. Yang, Biotechnol. Bioeng. 112(6), 1200–1209 (2015).
- Y. Fan, I. Jimenez Del Val, C. Müller, J.W. Sen, S.K. Rasmussen, C. Kontoravdi, D. Weilguny, and M.R. Andersen, Biotechnol. Bioeng. 112(10), 2172–2184 (2015).
- J. Liu, J. Wang, L. Fan, X. Chen, and D. Hu, World J. Microbiol. Biotechnol. 31(7), 1147–1156 (2015).
- M.M, St. Amand, D. Radhakrishnan, A.S. Robinson, and A.B. Ogunnaike, Biotechnol. Bioeng.111(10), 1957–1970 (2014).
- E. Pacis, M. Yu, J. Autsen, R. Bayer, and F. Li, Biotechnol. Bioeng. 108(10), 2348–2358 (2011).
- T. Surve and M. Gadgil, Biotechnol. Progr.31(2), 460–467 (2015).
- T. Tharmalingam, C.H. Wu, S. Callahan, and C.T. Goudar, Biotechnol. Bioeng.112(6), 1146–1154 (2015).
- C.K. Crowell, G.E. Grampp, G.N. Rogers, J. Miller, and R.I. Scheinman, Biotechnol. Bioeng. 96(3), 538–549 (2007).
- R.K. Grainger and D.C. James, Biotechnol. Bioeng. 110(11), 2970–2983 (2013).
- S. Kishishita, T. Nishikawa, Y. Shinoda, H. Nagashima, H. Okamoto, S. Takuma, and H. Aoyagi, J. Biosci. Bioeng. 119(6), 700–705 (2015).
- J.-M. Yang, J. Ai, Y. Bao, Z. Yuan, Y. Qin, Y.-W. Xie, D. Tao, D. Fu, and Y. Peng, Anal. Biochem. 448, 82–91 (2014).
- C.D. Agarabi, J.E. Schiel, S.C. Lute, B.K. Chavez, M.T. Boyne, K.A. Brorson, M.A. Khan, and E.K. Read, J. Pharm. Sci. 104(6), 1919–1928 (2015).
- S.N. Sou, C. Sellick, K. Lee, A. Mason, S. Kyriakopoulos, K.M. Polizzi, and C. Kontoravdi, Biotechnol. Bioeng. 112(6), 1165–1176 (2015).
- J.P. Kunkel, D.C.H. Jan, J.C. Jamieson, and M. Butler, J. Biotechnol. 62(1), 55–71 (1998).
- J.A. Serrato, L.a. Palomares, A. Meneses-Acosta, and O.T. Ramírez, Biotechnol. Bioeng. 88(2), 176–188 (2004).
- C.B. Michael, I.H.L. Daniel, and T.P. Eleftherios, Nat. Biotechnol. 11(6), 720–724 (1993).
- H. Aghamohseni, K. Ohadi, M. Spearman, N. Krahn, M. Moo-Young, J.M. Scharer, M. Butler, and H.M. Budman, J. Biotechnol. 186, 98–109 (2014).
- M. Yang and M. Butler, Biotechnol Bioeng. 68(4), 370–380 (2000).
- T.K. Ha, Y.G. Kim, and G.M. Lee, Biotechnol. Bioeng. 112(8), 1583–1593 (2015).
- J.K. Hong, S.M. Cho, and S.K. Yoon, Appl. Microbiol. Biotechnol. 88(4), 869–876 (2010).
- M. Yang and M. Butler, Biotechnol. Prog. 18(1), 129–138 (2002).
- S. Dietmair, M.P. Hodson, L.E. Quek, N.E. Timmins, P. Gray and L.K. Nielsen, PLoS One109(6), 1404–1414 (2012).
- D.C. Andersen and C.F. Goochee, Biotechnol. Bioeng. 47(1), 96–105 (1995).
- P. Olivova, W. Chen, A.B. Chakraborty, and J.C. Gebler, Rapid. Commun. Mass Spectrom.22(1), 22–29 (2008).
- A. Lim, A. Reed-Bogan, and B.J. Harmon, Anal. Biochem. 375(2), 163–172 (2008).
- S. Suzuki, Anal. Sci.29(12), 1117–1128 (2013).
- H. Geyer, and R. Geyer, Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 1764(12), 1853–1869 (2006).
- K. Mariño, J. Bones, J. J. Kattla, and P.M. Rudd, Nat. chem. Biol. 6
- S. Suzuki, T. Fujimori, and M. Yodoshi, Anal. Biochem. 354(1), 94–103 (2006).
- W.R. Alley Jr., B.F. Mann, and M.V. Novotny, Chem. Rev. 113(4), 2668–2732 (2013).
- M. Pabst, D. Kolarich, G. Poltl, T. Dalik, G. Lubec, A. Hofinger, and F. Altmann, Anal. Biochem. 384(2), 263–273 (2009).
- J.L. Desantos-Garcia, S.I. Khalil, A. Hussein, Y. Hu, and Y. Mechref, Electrophoresis32(24), 3516–3525 (2011).
- W.R. Alley Jr., M. Madera, Y. Mechref, and M.V. Novotny, Anal. Chem.82(12), 5095–5106 (2010).
- L.R. Ruhaak, G. Zauner, C. Huhn, C. Bruggink, A.M. Deelder, and M. Wuhrer, Anal. Bioanal. Chem.397(8), 3457–3481 (2010).
- Y. Hu and Y. Mechref, Electrophoresis33(12), 1768–1777 (2012).
- M.J. Oh, S. Hua, B.J. Kim, H.N. Jeong, S.H. Jeong, R. Grimm, J.S. Yoo, and H.J. An, Bioanalysis5(5), 545–559 (2013).
- M.D. Zolodz, J.T. Herberg, H.E. Narepekha, E. Raleigh, M.R. Farber, R.L. Dufield, and D.M. Boyle, J. Chromatogr. A1217(2), 225–234 (2010).
- M. Pabst and F. Altmann, Proteomics11(4), 631–643 (2011).
- K. Sandra, I. Vandenheede, and P. Sandra, J. Chromatogr. A1335, 81–103 (2014).
- J. Bones, N. McLoughlin, M. Hilliard, K. Wynne, B.L. Karger, and P.M. Rudd, Anal. Chem.83(11), 4154–4162 (2011).
- S. Hua, M.J. Oh, S. Ozcan, Y.S. Seo, R. Grimm, and H.J. An, TrAC, Trends Anal. Chem.68, 18–27 (2015).
- K. Baker, S. Flatman, and J. Birch, “Product Characterization from Gene to Therapeutic Product,” in Medicines from Animal Cell Culture, G. Stacey and J. Davis, Eds. (John Wiley & Sons, 2007), Chap. 22.
- D.A.M. Pais, M.J.T. Carrondo, P.M. Alves, and A.P. Teixeira, Curr. Opin. Biotechnol.30, 161–167 (2014).
- R. Korke, M. De Leon Gatti, A.L. Yin Lau, J.W. Eng Lim, T.K. Seow, M.C. Ming Chung, and W.S. Hu, J. Biotechnol. 107(1), 1–17 (2004).
- M.A. Bynum, H. Yin, K. Felts, Y.M. Lee, C.R. Monell, and K. Killeen, Anal. Chem. 81(21), 8818–8825 (2009).
- M. Doherty, J. Bones, N. McLoughlin, J.E. Telford, B. Harmon, M.R. DeFelippis, and P.M. Rudd, Anal. Biochem. 442(1), 10–18 (2013).
- M. Pucic, A. Knezevic, J. Vidic, B. Adamczyk, M. Novokmet, O. Polasek, O. Gornik, S. Supraha-Goreta, M.R. Wormald, I. Redzic, H. Campbell, A. Wright, N.D. Hastie, J.F. Wilson, I. Rudan, M. Wuhrer, P.M. Rudd, D. Josic, and G. Lauc, Mol. Cell. Proteomics10(10), M111–010090 (2011).
- D. Reusch, M. Haberger, M.H.J. Selman, P. Bulau, A.M. Deelder, M. Wuhrer, and N. Engler, Anal. Biochem. 432(2), 82–89 (2013).
- M.M. St. Amand, K. Tran, D. Radhakrishnan, A.S. Robinson, and B.A. Ogunnaike, PLoS One9(2), e87973 (2014).
- A.S Rathore, S.K. Singh, M. Pathak, E.K. Read, K.A. Brorson, C.D. Agarabi, and M. Khan, Biotechnol. Prog., in press (2015).
- A. Green and J. Glassey, J. Chem. Technol. Biotechnol. 90(2), 303–313 (2015).
Gunjan Narula is a project fellow in the Department of Chemical Engineering at the Indian Institute of Technology in New Delhi, India.
Vishwanath Hebbi is a graduate student in the Department of Chemical Engineering at the Indian Institute of Technology.
Sumit Kumar Singh is a graduate student in the Department of Chemical Engineering at the Indian Institute of Technology.
Anurag S. Rathore is a professor in the Department of Chemical Engineering at the Indian Institute of Technology in Delhi, India.
Ira S. Krull is a Professor Emeritus with the Department of Chemistry and Chemical Biology at Northeastern University in Boston, Massachusetts, and a member of LCGC’s editorial advisory board.
Articles in this issue
almost 10 years ago
Thermal Desorption Samplingalmost 10 years ago
Surfactant-Mediated Extractions, Part I: Cloud-Point Extractionalmost 10 years ago
What Quadrupole GC–MS Operators Need to Knowalmost 10 years ago
Monitoring Drug Behavior Using High Performance Affinity Chromatographyalmost 10 years ago
Vol 34 No 1 LCGC North America January 2016 Regular Issue PDFNewsletter
Join the global community of analytical scientists who trust LCGC for insights on the latest techniques, trends, and expert solutions in chromatography.
Advertisement
Related Content
Advertisement

Advertisement
Advertisement
Trending on LCGC International
1
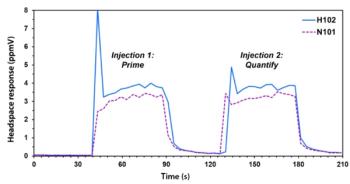
High-throughput Headspace Analysis of Volatile Nitrosamines and their Secondary Amine Precursors
2
Best of the Week: The Scientific Method in the Age of AI, Rewiring the Fundamentals
3
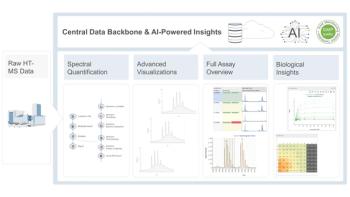
Unlocking Discovery Data: Why a Digital Ecosystem Matters for HT-MS
4
Detecting Estrogen Signatures in MCN Tumors via Targeted LC-MS/MS
5
