|Articles|April 1, 2018
- LCGC North America-04-01-2018
- Volume 36
- Issue 4
Peak Purity in Liquid Chromatography, Part II: Potential of Curve Resolution Techniques

In part II of this series, we explore how curve resolution methods advance purity assessments and illustrate one of the most popular techniques, which adds a powerful tool to the chromatographer’s arsenal.
Advertisement
Is that peak "pure"? How do I know if there might be something hiding under there?
In part I of this series we discussed how the peak purity tools commonly provided in chromatographic data system software could aid in the detection of impurities in liquid chromatographic analysis (1). Here, we go one step further, and explore how a class of chemometric techniques known as curve resolution methods can be used to differentiate between a target compound and impurities, and subsequently quantify them, even when their peaks are overlapped.
As in the previous installment (1), we focus on diode-array detection in liquid chromatography (LC–DAD). While mass spectrometric detection undoubtedly gives more selective information in the vast majority of cases, it is clearly a more complex detection mode and is prone to effects that can hamper quantitation such as ionization suppression because of matrix effects. The potential for highly precise quantitation of low-level impurities using DAD data is actually quite good, provided the spectra of the impurities have significantly different spectroscopic signatures as compared to the main peak. The latter point is of course an important caveat.
Multivariate Curve Resolution-Alternating Least Squares
In part I of this series we discussed the power of utilizing all of the absorbance information provided by a diode-array detector at multiple wavelengths to assess peak purity (1). Chemometric curve resolution techniques take this one step further. These techniques analyze the matrix of absorbance measurements at all wavelengths (that is, spectra) at all time points across a given time region of the chromatogram. Using a regression-based approach to determine how the spectra change over time, any impurities cannot only be discovered, but also be mathematically resolved from the target peak.
Here we illustrate one of the most popular curve resolution techniques, known as multivariate curve resolution-alternating least squares (MCR-ALS) (2–6). The basis for this technique is a multicomponent formulation of Beer's law given as
where Aλ represents the measured absorbance of a mixture solution at wavelength λ, b is the detection pathlength, ελ,X and ελ,Y represent the molar absorptivities at this wavelength for two chemical species X and Y, and cX and cY represent the concentrations of these species in the solution. For a two-component mixture, if absorbance measurements are obtained at two different wavelengths, and the molar absorptivities are known, it is possible to solve for the concentrations of the two species, X and Y, in the mixture solution via simple algebra. If measurements at more than two wavelengths are available, least squares regression is needed to obtain the concentrations. It is important to note that the assumption that the two (or more) signals are linearly additive is only valid in cases where the total signal is within the linear range of the detector (for example, at signals less than about 1500 mAU with DAD).
At this point, we generalize the discussion to a measurement x, and consider this as a signal in an LC–DAD chromatogram, such that the variable xi,j refers to the absorbance at the ith time point and jth wavelength of the chromatogram. Additionally, we consider the possibility that more than two chemical species may be present in the sample within the chromatographic peak, which gives the following expression:
Here, ci,n refers to the concentration of species n at the ith time point in the chromatogram, and sn,j refers to the molar absorptivity-pathlength product for species n at the jth wavelength. The full spectrochromatogram can be easily understood in terms of a matrix product. In matrix notation, equation 2 is commonly written as
where the rows and columns of matrix X represent the absorbance at each wavelength and time point, respectively, and the superscript T refers to the matrix transpose. This concept is illustrated schematically in Figure 1. If the molar absorptivities are known at all measured wavelengths for all species present in the peak, then it is straightforward to solve for the resolved chromatograms, C, as follows:
where the superscript † indicates the pseudo inverse operation. Equation 4 is simply a linear regression equation in matrix format. The columns of C are the individual component chromatograms (that is, each compound plus any background contributions), and the rows of ST are the individual component spectra.
Figure 1: Schematic for resolution of a spectrochromatogram represented by a matrix, X, into two component chromatograms and spectra contained by matrices C and S, respectively.
While in theory this approach could be a means of resolving overlapped chromatographic peaks, if there are unknown impurities present or uncharacterized mobile phase background components or species, then we do not have enough information to specify the S matrix. The MCR-ALS technique then becomes quite useful in this regard. Rather than exactly specifying S, an initial estimate for S is provided to the regression. This initial estimate can be obtained in a number of different ways. Pure variable methods are frequently used for this purpose. These methods seek to find the N most different spectra from the chromatographic data matrix, X, where N is the number of components needed to describe the measured data. The principle is that the most different spectra in the matrix are likely to be similar to the underlying pure component spectra. The caveat is that the number of components must be set by the user. Methods have been proposed for selecting the correct number of components such as scree plots; however, the only reliable method is evaluation of the results for multiple values of N. For a simple impurity screen, running MCR-ALS with two and three components to start should suffice, as one component would represent background, one would represent the target analyte, and if a third component is necessary, it is most likely because of an impurity peak.
Once this estimate for S is obtained, equation 4 is used to solve for the chromatographic profile matrix, C. Because the matrix S is only an approximation, C will only be an approximation as well. MCR-ALS can be considered an optimization method in which these C and S matrices are continuously improved with the goal of accurately representing the true underlying chromatographic and spectral profiles of each component. The power of MCR-ALS lies in the judicious implementation of constraints on the C matrix (and in subsequent steps, the S matrix as well) during this optimization. One frequently applied constraint is non-negativity, which allows the user to force the chromatographic profiles contained in C to have only positive values (6,7). Another constraint is unimodality, which forces each individual species chromatogram to exhibit a single peak (7). Many other constraints have been developed for MCR-ALS, but they are too numerous to describe here. Once C is constrained appropriately, the spectral matrix is updated via linear regression using equation 5:
Now, constraints can be applied to this S matrix as well; non-negativity is frequently used in this case too. By updating the S and C matrices in an alternating fashion (that is, equations 4 and 5), interspersed with the application of constraints, the final solutions for C and S will contain the pure component profiles of the individual chemical species within the chromatographic peak.
Application of MCR-ALS
We illustrate this approach using the chromatographic peak that was analyzed in part I of this series (1). Figure 2a shows the chromatographic peak, and Figure 2b shows the contour plot of the matrix X. We first applied a pure variable method (in this case the pure method in the Barcelona MCR-ALS toolbox, based on the SIMPLISMA algorithm [8–10]), and selected the three most different spectra within the spectrochromatogram. The corresponding time points are shown as circles in Figure 2a, and the three spectra at these points are shown in Figure 2c. It is likely that the spectrum shown in green represents a background spectrum, because it corresponds to a spectrum appearing in the baseline (green circle at 9.77 min in Figure 2a). After these initial estimate spectra are submitted to MCR-ALS, it should allow the algorithm to estimate the background contribution to the data, as well as the chromatographic peaks for each chemical species present within the profile.
Figure 2: (a) Chromatogram of impure peak at 212 nm; (b) representation of this chromatogram as a contour plot where the y-axis is the UV-visible absorbance spectrum axis and the x-axis is the chromatographic time axis; (c) three most "pure" spectra within the spectrochromatogram found at the points circled in (a).
The results for MCR-ALS analysis of this peak using these spectra for initial estimates are shown in Figure 3. Two peak shape responses within the chromatogram are resolved as shown in Figure 3a. These are two of the components contained in the matrix C, corresponding to two chemical species (peaks shown in blue and red), and a background contribution from the mobile-phase gradient shown in green. The normalized spectra contained in matrix S, which correspond to these species or contributions, are shown in Figure 3b. Note that the non-negativity constraint has been applied to the components corresponding to the real chemical species (shown in red and blue), while the background component (green) was not constrained. This flexible application of constraints leads to a powerful algorithm for curve resolution.
Figure 3: MCR-ALS results from the chromatogram shown in Figure 1. (a) Resolved pure component chromatograms; (b) resolved pure component spectra. The red and blue curves represent chemical species and the green curves represent background contributions.
Quantitation with MCR-ALS
A natural limitation of the MCR-ALS algorithm in this case is that there generally are multiple mathematical solutions that satisfy equation 3. Constraints are used to limit the possible solutions, but this generally does not provide a unique, chemically valid solution, especially when using MCR-ALS to analyze a single chromatogram, as described above. An extension of the MCR-ALS technique to analyze multiple chromatograms simultaneously is quite powerful in this regard, especially for quantitative analysis. In this approach, the analyst runs a series of calibration sample mixtures with varying concentrations of the target analytes, and obtains chromatograms for test samples with unknown concentrations of the target analytes. Because MCR-ALS resolves signals resulting from individual chemical species, these calibration solutes are not required to be individual standards and can, in fact, be mixtures of the compounds of interest, minimizing the number of calibration samples that need to be analyzed. These measured spectrochromatograms are appended together along the time axis to form an augmented matrix X as follows:
where the Xc are the L calibration chromatograms and the Xu are the M unknown chromatograms. MCR-ALS is carried out similarly to the approach described above. The resulting S matrix still consists of the N spectra of the pure component species, but the resulting C matrix now consists of L + M resolved chromatograms for each of the N species, appended together similarly as shown in equation 6. The resolved chromatograms and spectra for a dataset of five calibration standards, C1–C5, and one unknown, U1, are shown in Figure 4 (that is, L = 5; M = 1). The table above the figure shows the known concentrations of the standard mixtures, and it can be seen that the scaled peak intensities in the chromatograms (Figure 4a) are proportional to these concentrations. By integrating these resolved chromatographic peaks, calibration curves can be constructed, as shown in Figure 5.
Figure 4: (a) Resolved chromatograms for five calibration mixtures (C1 through C5) containing psoralen and angelicin; table shows the corresponding concentrations; (b) Resolved spectra for psoralen (red), angelicin (blue) and a gradient background contribution (green).
A clear advantage to handling multiple chromatograms simultaneously is that calibration information and estimates of unknown concentrations can be obtained very efficiently. Another advantage is the potential to add additional constraints to the analysis, which further limits the possible solutions for C and S. For example, if a blank chromatogram is included in the data set, the contributions of the chemical species for this chromatogram can be set to zero forcing the blank to be modeled using only the background components. Additionally, calibration constraints can be added to the analysis, which constrain the peak areas for the calibrated samples to follow an expected relationship between detector signal and concentration (11–13).
Figure 5: Calibration curves for (a) psoralen and (b) angelicin from MCR-ALS results. Colored circles indicate calibration points; black squares denote unknown sample points.
Of particular note here is the fact that two compounds present in the unknown sample have been reliably quantified, despite the resolution between the two peaks being significantly less than 1, and a high degree of similarity between their spectra. Here the chromatographic resolution of the two peaks is approximately 0.6.
Peak Capacity Enhancements via MCR-ALS
The performance of the MCR-ALS algorithm is highly dependent on the similarity of the spectra of the species contributing to the overlapped peak, as well as the signal-to-noise ratio (S/N) of the peaks. Here the similarity of the spectra for the two analytes psoralen and angelicin can be expressed by the correlation coefficient, which is 0.98 (see part I for further discussion).
The improvement of effective chromatographic performance can be quantified in terms of the peak capacity of the separation. The peak capacity of a gradient separation, nc, can be estimated as follows:
where tgrad is the time of the gradient, and wb is the average width of the peaks at the base. The Rs' term is the resolution required for effective quantitative analysis (14). Typically, chromatographers use an Rs' value of 1 when calculating peak capacity. Clearly, if peaks can be quantified at a resolution of less than 1 using curve resolution as discussed above, then the effective peak capacity has been increased. In recent work, we have developed a quantitative relationship between peak capacity and the signal-to-noise ratio of neighboring peaks and spectral similarity as measured by correlation coefficient. As an example, if the correlation coefficient between the overlapped spectra is 0.89 and S/N is 50, the chromatographic resolution required for quantitation is Rs' = 0.3. This results in a roughly threefold improvement in peak capacity relative to conventional use of DAD where the only means of separation is that provided by the column itself. Clearly, MCR-ALS can provide a significant enhancement in chromatographic method performance.
Availability of MCR-ALS in Software Packages
One hurdle to widespread usage of MCR-ALS is the lack of implementation of curve resolution options in commercial chromatographic data systems. Although commercial data systems for spectroscopy instruments (for example, infrared) frequently provide MCR-ALS or related curve resolution tools within their software, this situation is as not common for chromatographic data systems. To the best of our knowledge, only Shimadzu has recently added this capability to its data system software (15). The other option for chromatographers wishing to apply these methods to their data is to use one of the many available MCR-ALS toolboxes available for use in the Matlab programming environment. Eigenvector Research, Inc. sells its PLS Toolbox package, which includes MCR-ALS (16). Matlab toolboxes are freely available from the Barcelona MCR-ALS group (10,17) and the Olivieri group (18), with the latter toolbox specifically focused on calibration applications. The Olivieri and Barcelona MCR-ALS toolboxes are also available for users without access to Matlab through a stand-alone graphical user interface (17,18). There is also an ALS package available for the open-source R statistical software environment (19).
Because of the lack of integration with instrumental software, an extra step is required to export the raw spectrochromatogram and read it into the third-party software packages listed above. Unfortunately, this approach is not always straightforward, depending on the instrument software. Although a few extra minutes may be required to move the data and to analyze with the third-party software, it will often require less time than it would take to analyze samples using different chromatographic columns or to vary other method parameters to resolve impurity peaks and increase confidence that none are present.
Concluding Remarks
To those of us who have utilized MCR-ALS for chromatographic analyses, it is clear that this technique adds a powerful tool to the chromatographer's arsenal. While the peak purity approaches described in part I of this series can identify whether impurities are present, MCR-ALS can resolve the pure chromatographic profile, allowing quantitation of the target analyte and the impurity if standards are available for the compound. As mentioned earlier, MCR-ALS does require that compound spectra be at least slightly different; however, MCR-ALS is able to distinguish compounds with even small differences in spectra given a large enough S/N as shown in Figure 3.
Here we have limited our discussion to impurity analysis in LC–DAD; however, it is worth noting that MCR-ALS finds use in many other analyses such as metabolomics and environmental analyses as well as other instrumental techniques from hyperspectral imaging to LC with mass spectrometric detection to two-dimensional liquid chromatography (3,4,20,21). The latter will be the focus of the next installment in this series where we will look at how the additional separation dimension can help in the quest to determine peak purity particularly when spectrally indistinguishable impurities are present.
References
(1) S.C. Rutan, C.J. Venkatramani, and D.R. Stoll, LCGC North Am. 36(2), 100–110 (2018).
(2) A. de Juan and R. Tauler, Crit. Rev. Anal. Chem. 36, 163–176 (2006).
(3) A. de Juan, J. Jaumot, and R. Tauler, Anal. Methods 6, 4964–4976 (2014).
(4) D.W. Cook, M.L. Burnham, D.C. Harmes, D.R. Stoll, and S.C. Rutan, Anal. Chim. Acta. 961, 49–58 (2017).
(5) S.C. Rutan, A. De Juan, and R. Tauler, in: Comprehensive Chemometrics, S. Brown, R. Tauler, and B. Walczak, Eds. (Elsevier, Oxford, 2009), pp. 249–259.
(6) A. de Juan, S.C. Rutan, and R. Tauler, in: Comprehensive Chemometrics, S. Brown, R. Tauler, and B. Walczak, Eds. (Elsevier, Oxford, 2009), pp. 325–344.
(7) A. de Juan, Y. Vander Heyden, R. Tauler, and D.L. Massart, Anal. Chim. Acta. 346, 307–318 (1997).
(8) W. Windig, Chemom. Intell. Lab. Syst. 36, 3–16 (1997).
(9) F.C. Sánchez and D.L. Massart, Anal. Chim. Acta. 298, 331–339 (1994).
(10) R. Tauler, A. de Juan, and J. Jaumot,
(11) G. Ahmadi, R. Tauler, and H. Abdollahi, Chemom. Intell. Lab. Syst. 142, 143–150 (2015).
(12) R.R. De Oliveira, K.M.G. De Lima, A. De Juan, and R. Tauler, Talanta125, 233–241 (2014).
(13) A.C. de O. Neves, R. Tauler, and K.M.G. de Lima, Anal. Chim. Acta. 937, 21–28 (2016).
(14) J.M. Davis and P.W. Carr, Anal. Chem. 81, 1198–1207 (2009).
(15) S. Arase, K. Horie, T. Kato, A. Noda, Y. Mito, M. Takahashi, and T. Yanagisawa, J. Chromatogr. A. 1469, 35–47 (2016).
(16)
(17) J. Jaumot, A. de Juan, and R. Tauler, Chemom. Intell. Lab. Syst.140, 1–12 (2015).
(18) S.J. Mazivila, S.A. Bortolato, and A.C. Olivieri, Chemom. Intell. Lab. Syst.173, 21–29 (2018).
(19) K.M. Mullen,
(20) D.W. Cook and S.C. Rutan, Anal. Chem. 89, 8405–8412 (2017).
(21) J.M. Prats-Montalbán, A. de Juan, and A. Ferrer, Chemom. Intell. Lab. Syst. 107, 1–23 (2011).
ABOUT THE AUTHORS
Dwight R. Stoll is the editor of "LC Troubleshooting." Stoll is an associate professor and co-chair of chemistry at Gustavus Adolphus College in St. Peter, Minnesota. His primary research focus is on the development of 2D-LC for both targeted and untargeted analyses. He has authored or coauthored more than 50 peer-reviewed publications and three book chapters in separation science and more than 100 conference presentations. He is also a member of LCGC's editorial advisory board. Direct correspondence to:
Sarah C. Rutan is a professor of chemistry at Virginia Commonwealth University (VCU), in Richmond, Virginia, where she has been on the faculty for 33 years. Her research spans a broad range of areas in analytical chemistry and chemometrics, and is currently focused on the development of chemometric methods for improving chromatographic analyses, especially comprehensive 2D chromatography. She has more than 100 publications and numerous presentations on these topics.
C.J. Venkatramani is a senior scientist at Genentech USA and has more than 15 years experience in the pharmaceutical industry. He was the key member of Genentech technical team instrumental in the successful launch of gRed's first small molecule Erivedge, leading from development to commercial. His areas of interest include multidimensional chromatography and ultratrace analysis of genotoxic impurities.
Daniel W. Cook is a post-doctoral fellow in the pharmaceutical engineering laboratory within the Chemical and Life Science Engineering Department at Virginia Commonwealth University in Richmond, Virginia. In this role, he serves as the primary analytical chemist for the research efforts of the Medicines for All Institute. He received his BS from Randolph-Macon College in 2011 and his PhD in 2016 from Virginia Commonwealth University for his work focusing on the development and application of chemometric techniques to chromatography, particularly comprehensive 2D-LC.
Articles in this issue
over 7 years ago
New Chromatography Columns and Accessories for 2018over 7 years ago
Making HILIC Work for You—Column Selectionover 7 years ago
Vol 36 No 4 LCGC North America April 2018 Regular Issue PDFNewsletter
Join the global community of analytical scientists who trust LCGC for insights on the latest techniques, trends, and expert solutions in chromatography.
Advertisement
Related Content
Advertisement


Advertisement
Advertisement
Trending on LCGC International
1
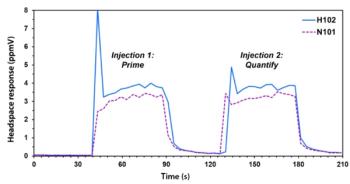
High-throughput Headspace Analysis of Volatile Nitrosamines and their Secondary Amine Precursors
2
Streamlined Method Development for Efficient and Reliable Lipid Nanoparticle Analysis
3
Best of the Week: The Scientific Method in the Age of AI, Rewiring the Fundamentals
4
Unlocking Discovery Data: Why a Digital Ecosystem Matters for HT-MS
5
