What Are Orphan Data?

The term orphan data is used frequently in the context of data integrity. What does it mean for chromatography data systems? How can we prevent or detect orphan data?
During a recent training course on data integrity, the question “What are orphan data?” was asked by one of the attendees. This gave me the excuse to write this column, but the real reason is that I forgot the column deadline. What we will cover here is understanding what orphan data means and how it applies to a chromatography data system (CDS). We will then look at ways to prevent the several types of orphan data being generated, starting with background information about the meaning and scope of raw data and complete data as they apply to chromatographic analysis. The aim of this column is to help laboratory reviewers, quality assurance (QA), and auditors to prevent or detect orphan data to ensure the integrity of results.
Understanding Raw Data and Complete Data
Raw data and complete data are Good Laboratory Practice (GLP) and Good Manufacturing Practice (GMP) terms used for records generated during a regulated activity. In an earlier “Questions of Quality” column, I discussed the definition and meaning of raw data in the context of a CDS (1). Raw data is a term that derives from FDA Good Laboratory Practice in 21 CFR 58.3(k) (2) published in 1978 and essentially means all data from original observations and activities, for example, acquisition, processing, interpretation, and calculation, to the reporting of the results. Most importantly, anyone should be able reconstruct the report of the work from the raw data (2,3). Please note, both the data integrity guidances from the MHRA (4) and PIC/S PI-041 (5) definitions of raw data are wrong as they only refer to original observations. The US GMP term complete data and the ability to equate this to raw data was discussed in my “Focus on Quality” column in Spectroscopy in 2016 (6).
Summarizing both raw data and complete data for a chromatographic analysis includes the following data: sample information, sample preparation records, acquisition method, instrument control file, CDS data files, processing method, calculations (unfortunately, I suspect, including any spreadsheets), system suitability testing (SST) parameters, audit trail entries, generation of the reportable result, plus instrument logbooks, laboratory notebooks, and worksheets. The principles outlined for CDS are applicable for other instrument systems.
As an evil auditor I want to be able to take any result and trace it back to the chromatography data, integration, and sample preparation records easily and transparently. I want to be able to trace all data from the start of the analysis to the result, including qualification data and tests performed after maintenance. In this latter review, I want to see all the data, including any problems encountered during the chromatographic work such as leaky liquid chromatography (LC) pump seals, blocked autosampler needles, clapped‑out column separations, and instrument failures—plus their resolution. All these problems will generate data that may or may not be usable for generating the reportable result. However, all data must be kept as they are part of raw data (2,3,7) or 21 CFR 211.194(a) complete data (8).
From Regulations to Procedures
GXP laboratories must interpret the applicable regulations and generate written instructions to perform tasks that generate records and reports (7).
This includes any work performed by service engineers and the associated agreements under EU GMP Chapter 7 (9). These instructions will include the requirements for recording or collecting data, processing them, and calculating the reportable results. They must be followed or a deviation must be raised. However, many of these standard operating procedures (SOPs) can be major works of artistic fiction as laboratory procedures must describe all working practices. Often procedures do not cover all activities, leading to poor records management practices or data falsification, or they can disguise activities that generate orphan data. If data integrity is compromised, the credibility of any laboratory is also compromised.
Definition of Terms
Now we need to focus on orphan data and we start with definitions of the two words and make the connection:
Orphan: A child whose parents have died
Data: Facts, figures, and statistics collected together for reference or analysis. All original records and true copies of original records, including source data and metadata and all subsequent transformations and reports of these data, that are generated or recorded at the time of the GXP activity and allow full and complete reconstruction and evaluation of the GXP activity (4).
At first sight, these two words make unlikely bedfellows. However, put on your chromatography glasses and look in more detail. The definition of data requires everything to be collected, while orphan indicates a separation or a break from the parents, in this case the main collection of data. Therefore, orphan data are separated from the data and results that are reported and purportedly represent the complete GXP record of the activity.
Orphan CDS Data
For a CDS, orphan data refer to:
- Data generated during a qualification or requalification of an instrument that is stored on a service engineer’s PC, or, if work has been repeated,
the selective reporting of passing results - During a chromatographic process, there are several areas where orphan data could be generated, for example, unofficial injections, multiple integrations, calculations, and short or aborted runs that are not reported or included as part of an official GXP record
- A run that has been excluded by dubious means, for example, unscientific invalidation of out-of-specification (OOS) results, or invalidation by manipulating SST injections to fall outside acceptance limits.
We will look at some of these types of orphan data and look at ways to identify or prevent them from occurring. Excluded from this discussion are copying data from one location to another to deliberately falsify a record and data deletions, for the reason outlined in the next section.
The reasons for orphan data may vary, from an analyst exercising “scientific judgement” (without reference to the regulations or laboratory SOPs) to deliberate falsification of data. Regardless of the reason, the dominoes fall thus: data integrity is compromised, followed by the quality of the decision made, and ultimately patients suffer.
Configuration of the CDS Application
One of the ways to restrict opportunities for generation of orphan data lies in the architecture and configuration of the CDS used by the laboratory. This will be an integral part of the validation of the system so that a reviewer’s or QA auditor’s job are made easier.
- The right system architecture: a CDS should be a networked system with an intelligently designed database. See the McDowall and Burgess four-part series on the ideal CDS for Regulated Laboratories for more information on this topic (10–13).
- Audit trails that cannot be turned off and are designed to monitor all changes to GMP records, including recording and highlighting all integration attempts automatically.
- A CDS application that saves all data automatically without having to prompt a user to do so.
- User roles and access privileges that avoid conflicts of interest.
Here it is important that no user has any privilege to delete data. - Single predefined data storage areas:
To avoid hiding orphan data and unofficial testing, it is critical that when performing an analysis that there is one, and only one, predefined data repository. - Use the system electronically and not as a hybrid: As many of you will ignore this, you will be left with more work to do.
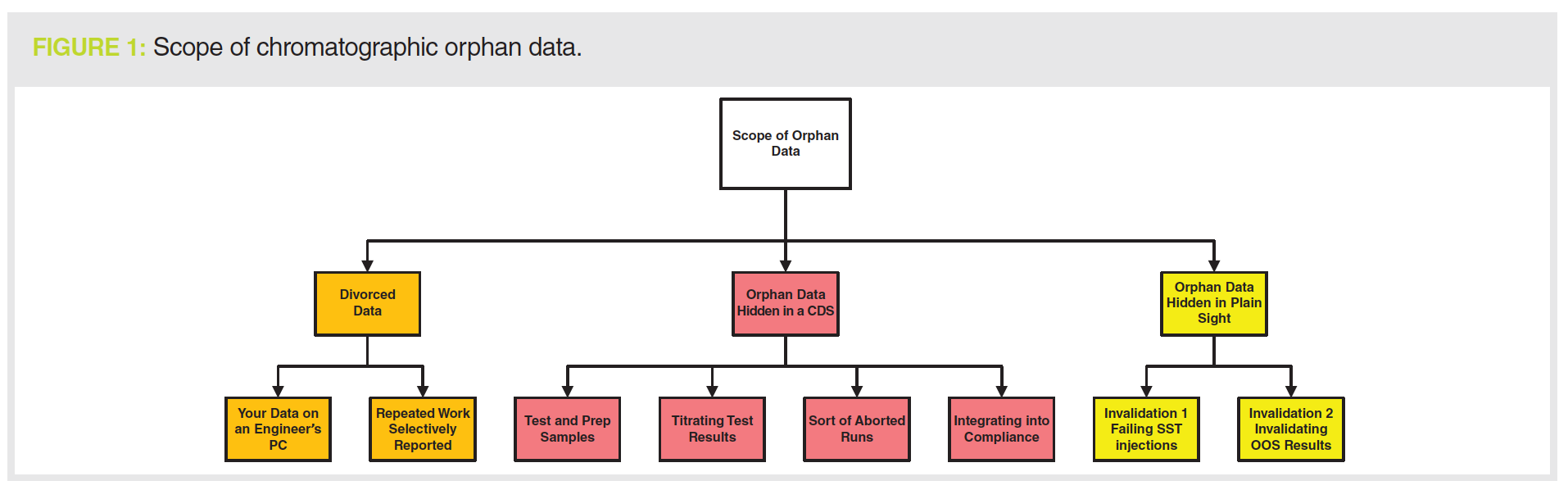
Orphan Data: Divorced, Hidden, or in Plain Sight?
We will look at the main means that service engineers and analysts use to generate orphan data and the ways that you can prevent or detect them in the sections below and in Figure 1. There are three types of orphan data that reviewers, QA, and auditors need to be aware of:
- Divorced data:
Your qualification data are stored on a service engineer’s laptop
Repeating qualification work and selectively reporting the passing result - Hidden in the CDS:
Test or prep injections
Titrating test results
Short or aborted runs
Integrating into compliance - Hidden in plain sight:
Invalidated SST injections
Invalidated OOS results.
We will start with divorced examples of CDS orphan data.

The Angel of Death Calls
Our first port of call focuses on a visit from the Angel of Death, sorry, the service engineer, who typically will perform a preventative maintenance visit and requalify your chromatographs. It depends on the level of paranoia in a company as to whether a service engineer has access to the CDS. If a laboratory is sensible, there is a user role called service or engineer where the access privileges for performing service and qualification are configured. The engineer(s) must be named CDS user(s) and the account enabled and disabled before and after the qualification visit. All qualification data are within the CDS or linked to any external tests such as temperature or flow using calibrated instruments so that an authorized laboratory user can access and review the data and work is attributed to a named individual.
If the engineer is denied CDS access, Pandora’s box is opened and the evils of the world unleashed. Actually, it is the engineer’s PC that is opened, but the end result is the same. After disconnecting the chromatograph from the CDS, the engineer connects their PC with the CDS software (Note: this is not testing the system under actual conditions of use). Depending on whether the original equipment manufacturer or a third party is used for service and requalification, you may or may not have qualification data in the same CDS as the one you use. The engineer will also be an administrator of this CDS, with an obvious conflict of interest and the right to hide and delete data or even change the PC clock. If data are stored on the engineer’s PC,
how many attempts have there been to get a chromatograph to pass a test?
After qualification is complete, you will get a printed report of work performed. However, there are a few potential problems:
- Have you approved the work prior to execution and do you understand it?
- Do you get sight of the electronic records generated by the qualification? If yes, have you reviewed them?
- Do you have copies of the electronic data? If yes, are they legible and can you understand them?
If you don’t have copies of the data, the engineer certainly does! But the individual has just ridden off into the sunset. How long do you think your data will survive before deletion?
Remember 21 CFR 211.194(d): Complete records shall be maintained of the periodic calibration of laboratory instruments, apparatus, gauges, and recording devices required by § 211.160(b)(4) (8). Complete records include the data from all attempts at qualification, any explanation of reasons for failure, how they were resolved, and all should be included in the qualification report.
If this is not achieved, your qualification data have divorced you. Let’s move on to the greener pastures of orphan data in sample testing
Test and Prep Samples
One classic example of orphan data are injections stored in directories called
Test and Prep, or in one example a directory called Not To Be Shown To FDA. These directories contained sample injections that were used to see if a batch would pass or not. Restricting where data files are stored will make unofficial testing more visible, but this results in the rise of the short, aborted, or incomplete runs that we will see in the next section.
Before we move on, we need to distinguish unofficial testing from system evaluation or column conditioning injections. The latter is allowed but only if you use reference standard injections and never sample solutions. You can see a discussion on this topic on the FDA website (14) and in a “Questions of Quality” column on peak integration (15). FDA require the practice of column conditioning to be traceable back to a method validation report and to be scientifically justified (14).
It is critical that if you use evaluation or conditioning injections that the practice must be documented in a SOP, and must make it clear that these injections are part of complete/raw data of the sequence and are evaluated for each run. I suggest having acceptance criteria for these injections to show that the system is ready. Also, be very careful in how you name these injections.
Titrating CDS Results
One way of generating passing results is to titrate the data to obtain a passing result by changing values in the sequence file, such as sample weight, reference standard purity, or water content. This was identified in one of the 2005 Able Laboratories 483 observations (16):
Observation 5: …. The substitution of data was performed by cutting and pasting of chromatograms, substituting vials, changing sample weights, and changing processing methods……
Sample weights were changed by the analyst until a passing result was obtained.
One way to prevent the generation of this type of orphan data is through effective searching of audit trail entries, but to do this effectively requires a CDS with functions to help. Ideally, a CDS should identify where modifications in a data set have been made so that the reviewer or auditor does not have to trawl through all entries. Instead, they can focus their effort where data are changed to meet the requirements of Annex 11 (17) and PIC/S PI-041 (5).
Short or Aborted Runs
Houston, we have a problem is another way of looking at this topic. A short run consisting of one or two injections that is then stopped or aborted, which could be seen as the updated version of Test and Prep injections. As there are no deletion rights in the CDS, what should an analyst do to explain these unofficial test data? Instrument failure—easy-peasy!
To highlight the issue, a recent citation from Aurobindo issued in August 2022 (18) showed the message centre of a CDS had logged 6337 (yes, really!) error messages from 1 July to 1 August 2022. Analysis of these identified the following entries:
- 411 instrument failure messages
- 13 messages of sequence stopped because of error or sequence stopped by user
- 20 Failed to get the newest information of the batch queue because of the communication failure messages.
These could be the start of potential instances of orphan data that must be investigated further.
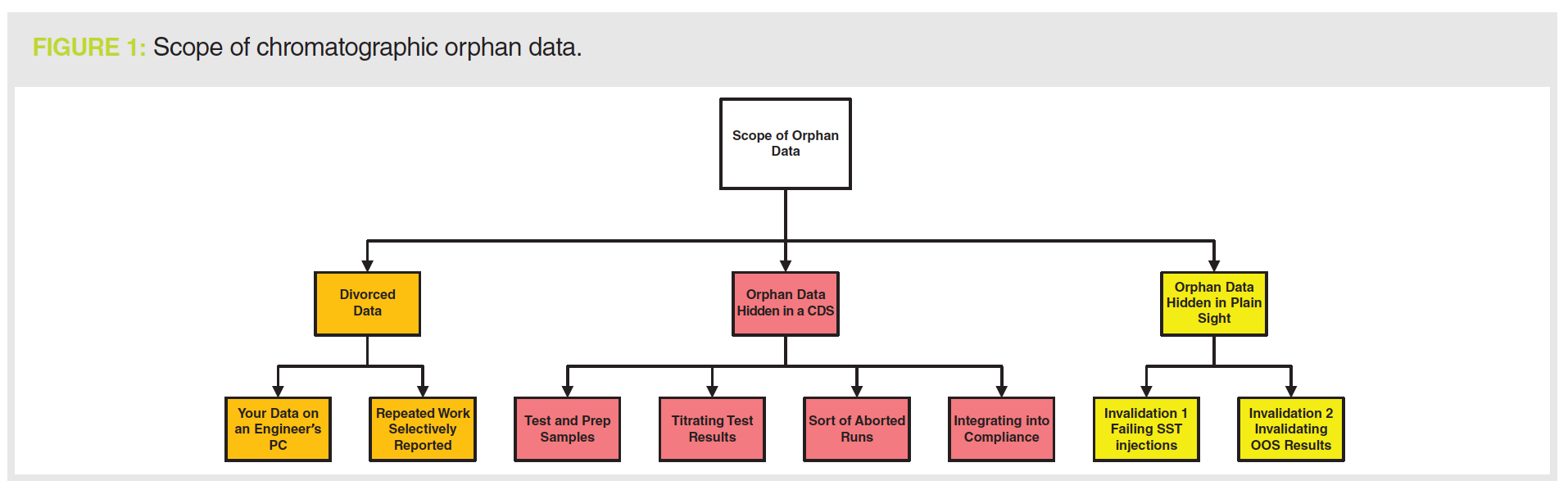
Enter stage left the unsung hero of data integrity—the instrument logbook! See 21 CFR 211.182 (8) and Chapter 4.31 (7) for the regulatory requirements that most people don’t understand and can’t implement.
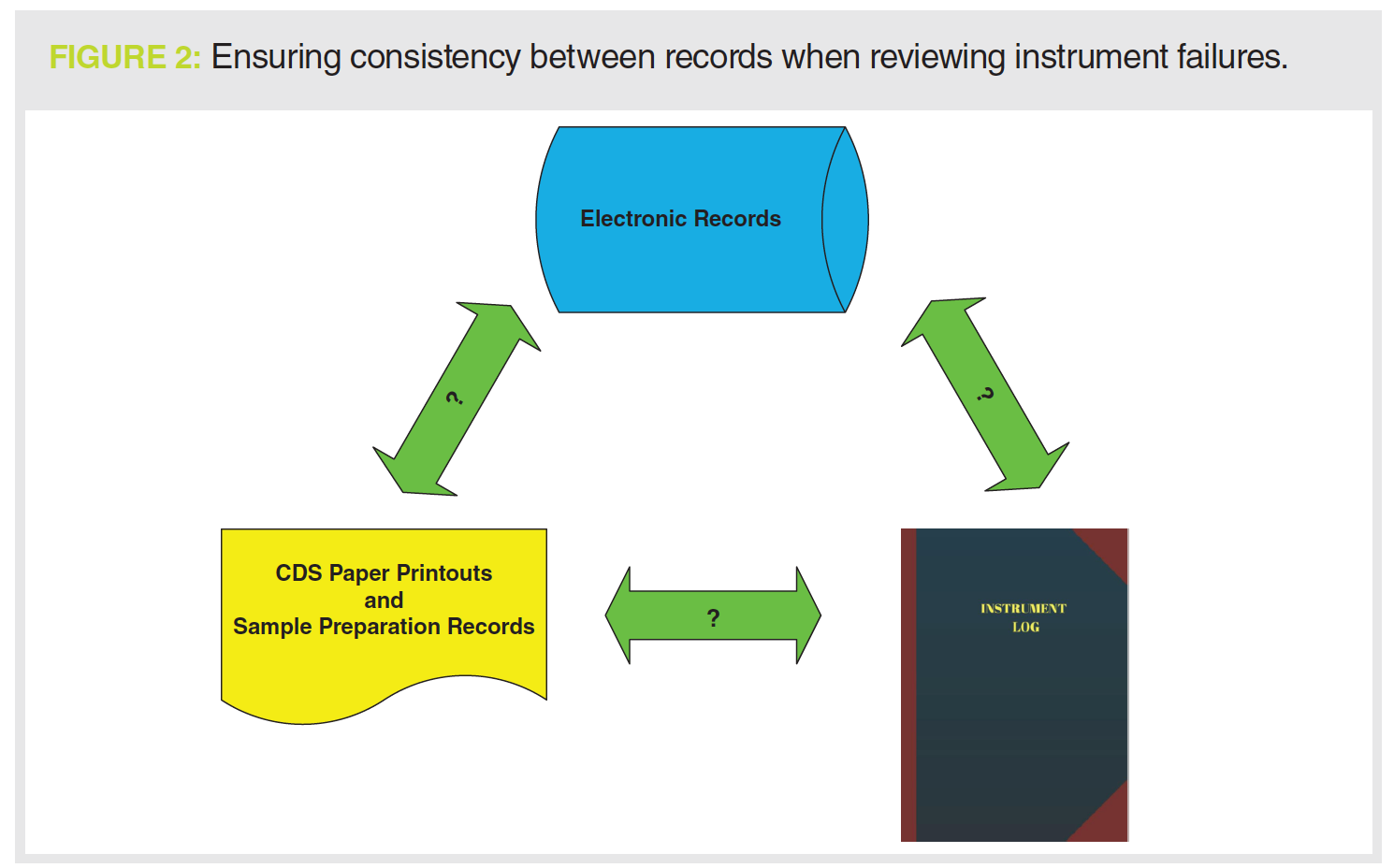
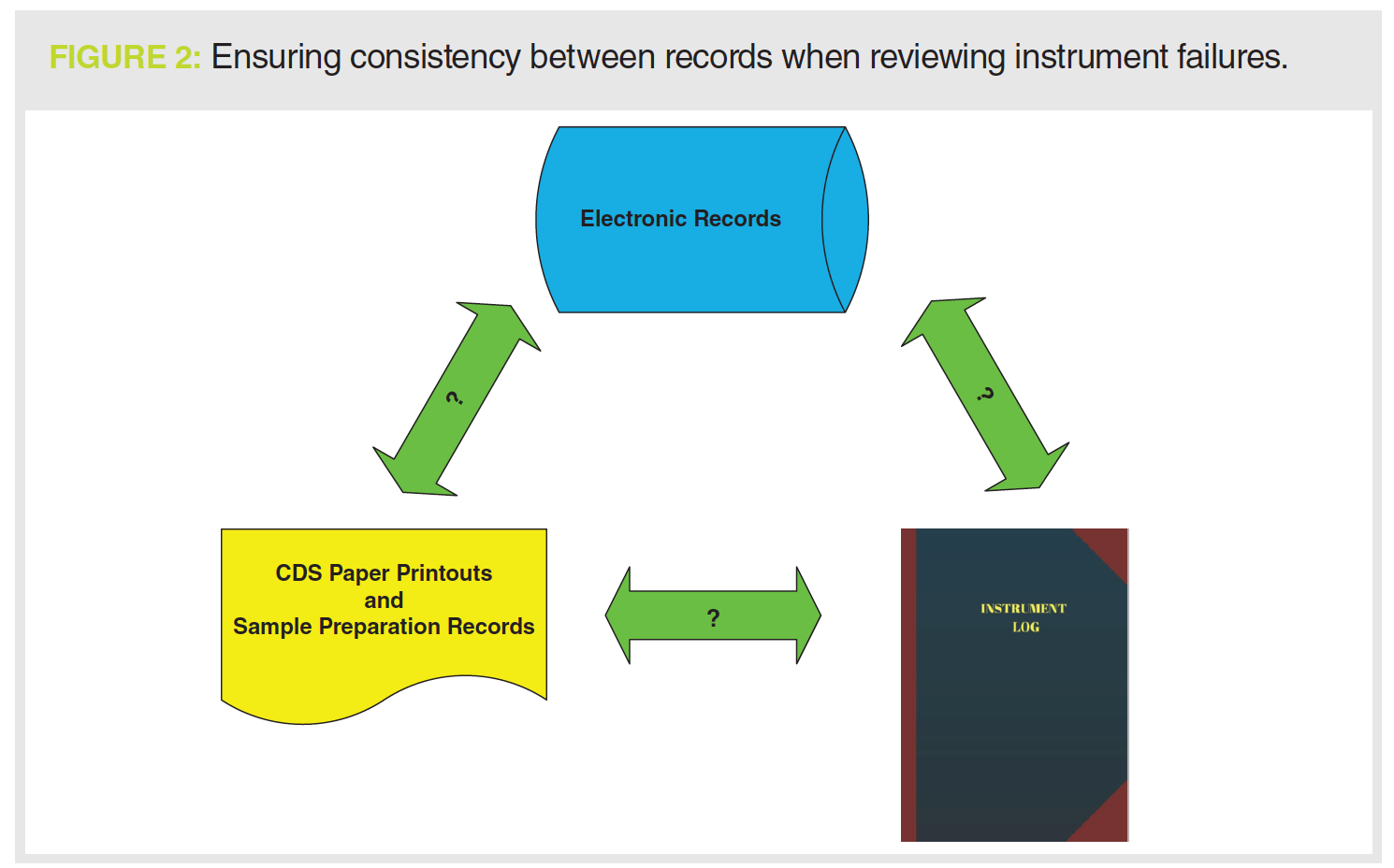
If there is an instrument failure, the first place to look is the instrument logbook to see what sort of failure and then at the chromatograms (both electronic and printout) to see how the failure is seen (see Figure 2). For example, to support a justification for an instrument failure with a pump seal leak, you should see the peak shape get worse and retention times get longer as mobile phase is no longer being pumped consistently at the set rate.
You should then assess what was done to fix the problem; back to the instrument logbook to see the maintenance entries. What about any requalification work? Is there a logbook entry for any testing that the leak was fixed? And where are the data or records associated with this work?

Integrating into Compliance
A more subtle type of orphan data is an analyst carrying out multiple peak integration attempts. In some CDS, they will be prompted to save each reintegration, meaning you only have the audit trail entries to assess. In other CDS applications, each attempt will be automatically saved, but only the “correct” result will be reported.
As indicated in multiple warning letters and 483 observations, peak integration must be controlled, otherwise the wrath of an inspector will descend. Peak integration has been covered in two “Focus on Quality” articles (15,19), where the sins of others were discussed extensively; however, peak integration 483 citations are still found, as seen in August 2022 with Sun Pharmaceuticals (20):
Observation 4: When an analyst processing chromatography data determines the existing processing method is not appropriate or chooses to use a different processing method for samples within the same sample set during impurities analysis, the original chromatogram is not saved. The analyst can integrate the chromatogram and see the results within the software, but does not save the result. To change the processing method the analyst must document that the integration was inappropriate and get approval on Form 000505. But the original chromatogram is not saved to justify the changes to the processing method were necessary.
This observation shows that the procedural controls are totally ineffective, as the CDS lacks the technical controls and it should have been replaced with a compliant application. This is not so much orphan data as no data.
Observation 5: Procedures to ensure accurate and consistent integration of chromatographic peaks have not been established. The analysts can choose the integration algorithm and manually enter timed integration events into the processing methods. Procedures have not been established to ensure the appropriate and consistent use of these timed integration events.
Welcome to the Wild West where anything goes with peak integration and you can generate large volumes of orphan data under the guise of scientific judgement. Regardless of the analysis, all integration must be automatic first time, every time. Then you can perform manual intervention for all injections, but only in specified cases can manual integration be used and the chromatographer can reposition the baselines (19).
SST Failures
FDA’s Christmas present for chromatographers is found in the guidance on investigating OOS test results issued in 2006 and 2022 (21,22). Requirements for SST results are found in <621> and 2.2.46 of the United States Pharmacopeia and European Pharmacopoeia (23,24), respectively, to ensure that a chromatographic system is fit for use. FDA states in Section IIIA (22):
Certain analytical methods have system suitability requirements, and systems not meeting these requirements should not be used. For example, in chromatographic systems, reference standard solutions may be injected at intervals throughout chromatographic runs to measure drift, noise, and repeatability. If reference standard responses indicate that the system is not functioning properly, all of the data collected during the suspect time period should be properly identified and should not be used. The cause of the malfunction should be identified and, if possible, corrected before a decision is made whether to use any data prior to the suspect period.
This is good scientific advice by FDA. However, it is also a gilt-edged invitation to unscrupulous chromatographers to adjust the SST integration to be out of acceptance limits with a failing batch. Thus, where a sequence fails due to SST failure examine the peak integration and audit trail entries of the
run and the SSTs injections specifically. This should be accompanied by a critical review of the laboratory investigation to ensure a scientifically sound reason for the invalidation. Don’t forget to review the instrument logbook as well, especially if the investigation cites instrument failure (see Figure 2).
Invalidating OOS Results
The last example of orphan data hiding in plain sight is invalidating an OOS result with an inadequate laboratory investigation. The problem with many laboratory investigations is that the outcome is “analyst error”, in which the data are invalidated, the analyst has their ears boxed, they are retrained, and all is well with the world. Until the next laboratory investigation…
Lupin received a 483 observation for invalidating 97% of stability OOS results (25) to avoid the need to file a field alert with FDA. As described above, critically evaluate the investigation and find what the assignable cause is and if it is scientifically sound. FDA is on the case with the quality metrics guidance where the only laboratory metric is percentage invalidated OOS results (26). Additionally, even where scientific judgement supports the decision, repeat failures for the same reason need to be investigated.
Common Issues
All of the orphan data examples discussed above are failures of one or more ALCOA+ or ++ criteria (27,28). Moreover, a laboratory has failed to put in place either the technical controls in the CDS and/or procedural controls and training to prevent orphan data being generated and detected by the second person reviewer, coupled with QA data integrity audits for oversight. For qualification of chromatographs, it is important that laboratories allow service engineers access to a CDS to avoid divorced data. One question arises: Do the current CDS applications have the functions available to help reviewers detect and examine cases of potential orphan data?
CDS Functions to Identify Orphan Data
To help reviewers, what new functions are required for a CDS? Two areas spring to mind:
- Automatic identification of short or aborted runs. This would be used in second-person review of data by a supervisor, but also during data integrity audits by QA.
- SST failures is another area where automatic identification would greatly assist review and audit.
Summary
We have discussed orphan CDS data and the ways that they can be generated to falsify results. A CDS with appropriate technical controls implemented and validated can prevent some of them occurring, but for the remainder reviewers and auditors need to be trained and remain vigilant to detect them. Further functions in CDS applications would also greatly help detection of orphan data.
Acknowledgements
I would like to thank Mahboubeh Lotfinia and Paul Smith for their helpful comments during the writing of this column.
References
- R.D. McDowall, LCGC Europe 30(2), 84–87 (2017).
- 21 CFR 58, Good Laboratory Practice for Non-Clinical Laboratory Studies (Food and Drug Administration, Washington, DC, USA, 1978).
- OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring Number 1, OECD Principles on Good Laboratory Practice (Organisation for Economic Co-operation and Development, Paris, France, 1998).
- MHRA, GXP Data Integrity Guidance and Definitions (Medicines and Healthcare Products Regulatory Agency, London, UK, 2018).
- PIC/S PI-041, Good Practices for Data Management and Integrity in Regulated GMP / GDP Environments Draft (Pharmaceutical Inspection Convention / Pharmaceutical Inspection Cooperation Scheme, Geneva, Switzerland, 2021).
- R.D. McDowall, Spectroscopy 31(11), 18–21 (2016).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 4 Documentation (European Commission, Brussels, Belgium, 2011).
- 21 CFR 211, Current Good Manufacturing Practice for Finished Pharmaceutical Products (Food and Drug Administration, Silver Spring, Maryland, USA, 2008).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 7 Outsourced Activities (European Commission, Brussels, Belgium, 2013).
- R.D. McDowall and C. Burgess, LCGC North America 33(8), 554–557 (2015).
- R.D. McDowall and C. Burgess, LCGC North America 33(10), 782–785 (2015).
- R.D. McDowall and C. Burgess, LCGC North America 33(12), 914–917 (2015).
- R.D. McDowall and C. Burgess, LCGC North America 34(2), 144–149 (2016).
- FDA, Questions and Answers on Current Good Manufacturing Practices—Laboratory Controls: Level 2 Guidance. Available from: https://www.fda.gov/drugs/guidances-drugs/questions-and-answers-current-good-manufacturing-practices-laboratory-controls
- H. Longden and R.D. McDowall, LCGC Europe 21(12), 641–651 (2019).
- Able Laboratories Form 483 Observations. 2005. Available from: https://www.fda.gov/media/70711/download
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 11 Computerized Systems (European Commission, Brussels, Belgium, 2011).
- Aurobindo Pharma Limited, Unit X1 FDA 483 Observation August 2022 (Food and Drug Administration, Silver Spring, Maryland, USA, 2022).
- R.D. McDowall, LCGC Europe 28(6), 336–342 (2015).
- Sun Pharmaceutical Industries Limited FDA 483 Observations, 2022.
- FDA, Guidance for Industry Out of Specification Results (Food and Drug Administration, Rockville, Maryland, USA, 2006).
- FDA, Guidance for Industry, Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production (Food and Drug Administration, Silver Spring, Maryland, USA, 2022).
- United States Pharmacopeia General Chapter <621> “Chromatography” (United States Pharmacopeial Convention, Rockville, Maryland, USA).
- European Pharmacopoeia, EP 2.2.46 Chromatographic Separation Techniques (European Pharmacopoeia, Strasbourg, France).
- FDA 483 Observations: Lupin Limited (Food and Drug Administration, Silver Spring, Maryland, USA, 2017).
- FDA, Guidance for Industry Submission of Quality Metrics Data, Revision 1 (Food and Drug Administration, Rockville, Maryland, USA, 2016).
- WHO, Technical Report Series No.996 Annex 5 Guidance on Good Data and Records Management Practices (World Health Organization, Geneva, Switzerland, 2016).
- R.D. McDowall, Spectroscopy 37(4), 13–19 (2022).
About The Column Editor
Bob McDowall is Director of R.D. McDowall Limited, Bromley, UK. He is also a member of LCGC Europe’s editorial advisory board. Direct correspondence to: amatheson@mjhlifesciences.com





 Workshop on Analytical Techniques and Implementation")




The LCGC Blog: Are You Sick of Hearing About ChatGPT?
August 15th 2023Are you interested by ChatGPT's vast potential? Or are you getting tired of reading or hearing about ChatGPT? If the latter, this blog post might give you some tips and tricks on taking advantage of ChatGPT while avoiding its application beyond its intended scope.



