
|Articles|February 1, 2021
- February 2021
- Volume 34
- Issue 2
What’s Good About the WHO Good Chromatography Practices Guidance? Part 2
Author(s)R.D. McDowall

Listen
0:00 / 0:00
Key Takeaways
- The WHO guidance on Good Chromatographic Practices lacks clarity, organization, and comprehensiveness, with key areas inadequately addressed or omitted.
- Misplaced clauses and vague language, with an over-reliance on "should" rather than "must" statements, undermine the document's effectiveness.
In September, the World Health Organization (WHO) issued a new guidance document on Good Chromatography Practices. What guidance does it contain and is it useful? Has the document failed its system suitability test (SST) acceptance criteria?
Advertisement
In September 2020, the WHO issued a guidance document on Good Chromatographic Practices (1). The scope, content, and sections of the document dealing with the system and its set up were discussed in Part 1 (2). In this part, we’ll take a close look at the sections dealing with chromatography analysis and see if any additional bouts of lalochezia occur.
Chromatographic Analysis
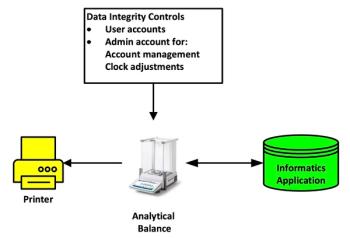
Let’s get down to some chromatography, as the remainder of the guidance document is focused on analysis of samples, as shown in Figure 1, and
has the following sections:
- Solvents, buffer solutions and mobile phases (Section 9)
- Column management (Section 10)
- Sample management and sample set (Section 11)
- Chromatographic methods (acquisition and processing) (Section 12)
- Peak integration (Section 13)
- Data management (Section 14)
I am at a loss to understand the difference between processing and peak integration in Sections 12 and 13 respectively? Especially as there is no mention of processing in Section 12!
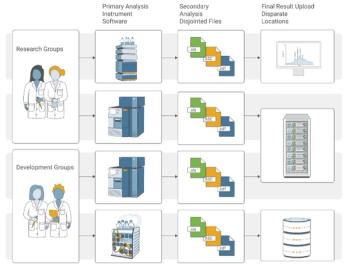
OK, competition time! What’s missing from the list above? Give up? Quite a lot! Figure 2 shows the contents of the GChromP document (green boxes) mapped against a generic analytical process (yellow boxes) with the main missing
tasks (red boxes). Presented in this way you can see the areas that the WHO guidance omits. The dashed lines indicate partial coverage in the WHO guidance document. There is no mention of the following that I believe should be part of a chromatography guidance to ensure data integrity:
- Analytical Procedure: The overall analytical procedure is the controlling document for the work and this is not mentioned here as such. This is not to be confused with the chromatographic method that is discussed partly in Section 11 and in more detail in Section 12 of this guidance as this only focuses on column management then the set up and processing within the chromatography data system (CDS). The difference between a procedure and method was discussed in a stimulus paper published in Pharmacopoeial Forum by Schofield et al. (3). Here the difference between procedure and method was stated as: the term analytical method refers to operational components comprised of instrumentation, reagents, standards, sample preparations, calibrations, controls, and suitability criteria on the system. ... the method is the “ruler” used to make measurements. A measurement is an output from a single implementation of the method on a sample of a test article. By contrast the term analytical procedure will refer to a use of the method to make a decision...for example, batch release. If the WHO guidance only wanted to focus
on the method why not say so? - Sampling Plan: A sampling plan as the guidance only starts when the sample arrives in the laboratory, but if the sample is not taken correctly then all the work in the laboratory is wasted. WHO have published detailed guidance for sampling with three suggested plans (4) but this is not referenced here.
- Sample Preparation: The phase where samples are prepared for injection into a chromatograph is not mentioned apart from 11.4. This is the most critical part of the analytical process after sampling and can be associated with the most experimental error. It is also mostly manual with paper records and is a key point for ensuring ALCOA+ criteria (attributable, legible, contemporaneous, original, and accurate, plus complete, consistent, enduring, and available) are met plus assessing if any data integrity lapses or falsification have occurred.
- Manual Data Entry: Manual entry of data into the CDS (for example, sample identities, weights, dilutions, and so on) are not mentioned. This a key area to check for transcription errors.
- Instrument Control: There is no mention of instrument control of the chromatograph, that is a feature of most chromatography data systems.
- Comparative Technique: There is no mention of the fact that chromatography is a comparative technique and that standards and samples should be integrated similarly to avoid biasing the results. It is essential that this is included in Section 13 on peak integration but is not.
- No Spreadsheets: Calculations should be performed within the CDS, not outside of it, to avoid transcribing and checking manual data entries.
- Second Person Review: No second person review of data generated is mentioned apart from one word: reviewed. This is a key requirement for ensuring the integrity of any laboratory result and the underlying records and data, and it is missing in action. Given its importance, there should be a separate section to detail the review tasks by a second person.
- Short or Aborted Runs: Injecting one or two sample injections is a current practice for unofficial testing, to see if the batch passes or not. This is an area that needs to be checked by the second person review as a minimum and as part of a quality assurance (QA) data integrity audit.
- OOS Results: Missing in action, but an investigation should follow the out of specification (OOS) results guidance from the FDA (5) this is a current area of testing into compliance by invalidating these results (6).
- Training: There is no mention of training in either data integrity or ethics, as well as chromatographic analysis, especially integration, anywhere in this document.
How much a guidance document goes into detail is at the discretion of the authors. However, if items are to be omitted from the scope of a guidance document then it is the responsibility of the authors to state this. In this respect the authors and quality oversight for the document have failed. Please excuse me while I have another bout of lalochezia.
Reagents and Column Management
Section 9 deals with solvents, buffers, and mobile phases and is relatively straightforward except for one phrase. Clause 9.1 covering the use of these items states in the last sentence: These should be used within appropriate, scientifically justifiable timelines. Why not simply say that expiry dates should be justified scientifically?
Section 10 covers column management and provides general advice about column management, from purchase to how to handle the column in the laboratory and keeping records of each one’s use. However, there should be a warning that if a column is used for different analytical methods, it may change the chromatographic properties over time. Ideally it is better to have dedicated columns, though cost may be a factor in some laboratories.
Clause 10.4, requiring chromatograph tubing and fittings to be appropriate (but you are left to define that!), is in the wrong place and should be in Section 3 on chromatographs. Given the organization of the document...
Clause 10.5 suggests that the only method of monitoring a column performance is to calculate the theoretical plates. In my experience, very few laboratories use plate count to measure column performance—it is an academic exercise because it depends on the separation in question. For example, if separation of two closely running peaks is required, then peak resolution is a better criterion. You could have a situation where the plate count is acceptable but peaks are not resolved. Perhaps a better way of expressing this is that the criteria for monitoring any column’s performance should be scientifically sound. This leaves the laboratory to select and monitor appropriate criteria through system suitability tests (SSTs). On the topic of SSTs, clause 4.8 mentions that on SST failure there should be a (corrective action and preventative action) CAPA generated. This is stupid. What must happen is that the failure must be documented, investigated, and appropriate action agreed which may include a CAPA plan but not necessarily so. Consider a pump leak as the source of SST failure: document, replace the seal, requalify, and get on with the analysis, there is no need for a CAPA. It raises the question: how many of the authors have actually worked in an analytical laboratory?
Sample Management and Preparation
Sample management is presented in overview but sampling and the sample plan is omitted, and so is reference to the overall analytical procedure. Notwithstanding, the outline of sample preparation and placing the vials in the correct order to match the sequence file order is presented well. However, from 11.9 to 11.13 we move into SST injections and analysis of the samples before discussing the chromatographic method in Section 12 and therefore these are in the wrong sequence (sorry!). Unfortunately, manual entry of sample information such as identity, lot number, weight, dilution factor, and standard purity, which can be a source of transcription errors, is not even considered.
Trial Injections
Clause 11.9 is badly phrased and states that trial or system check injections that are not specified as an injection sequence is not recommended. (Normally, only standard solutions may be used for this purpose, unless otherwise needed and justified (e.g. biologics). This is similar to the approach that Heather Longden and I discussed in a recent Questions of Quality on peak integration (7) but is written more clearly. There is a Level 2 guidance question and answer on the FDA website that goes into more detail about this topic by asking question 17: Is it ever appropriate to perform a “trial injection” of samples?
No. ..... This is in contrast to the appropriate practice where an injection of a standard is performed with the sole intention of determining if the chromatographic system is fit for purpose. The injection of trial samples is not acceptable, in part, because all data from analysis of product samples must be retained and reviewed...
Column conditioning does not involve injecting a sample from a lot and is not considered a trial injection. When its use is scientifically justified, column conditioning should be fully described in the method validation package as to the conditions needed to make the measurement (i.e., based on data from the method validation) and should be clearly defined in an approved and appropriate procedure. Only validated test methods that demonstrate accuracy, sensitivity, specificity, and reproducibility may be used to test drugs (21 CFR 211.165[e]). Consistent and unambiguous injection nomenclature should be used, and all data from the column conditioning, including audit trail data, should be maintained and subject to review (8).
Using standard injections to confirm that the column is conditioned is an acceptable practice BUT it needs to be scientifically justified, included in the method validation report and MUST be part of complete data for the second person review under 21 CFR 211.194(a), clause 8 (9). There are no excuses or justifications for taking any different approach.
Chromatographic Methods
Section 12 is titled Chromatographic Methods (acquisition and processing) but there is not much in this section about acquisition and nothing about data processing. The order of clauses is random and confusing (lucky dip time or a chat after a session at the bar?):
- 12.1: Chromatographic methods should be suitable for intended use with acceptance criteria, for example, selectivity, peak symmetry, repeatability, and integration conditions.
- 12.2: Non-pharmacopoeial methods should be validated.
- 12.3: Methods should be saved in the CDS by authorized personnel and should only be changed where justified.
- 12.4: CDS software should be validated! What!? This clause does not belong here it should be in Section 4! This clause is also shared with the statement that methods selected for acquisition and processing should be traceable and reflected in the audit trail—how about moving this to the audit trail section?
At this point in the document, even an extended bout of lalochezia fails to work and I’m getting depressed as I’ve still got two more sections to review.
Peak Integration
Hope springs eternal and I’m hopeful that I’ll find something positive to say about this part of the document. Clause 13.1 with its scientifically sound approach to integration raises my hopes, but they are dashed when I get to clause 13.2. Here I find that I should connect the chromatograph to a CDS—sorry—to computerized chromatographic data capturing and processing systems. What is this and why do we need to connect a chromatograph to two of them? Again, the document organization is appalling with an abject failure in technical understanding as well as editorial and quality oversight.
After discussing some of the types of integration and the ideal peak shape for integration (symmetrical) we come to manual integration. Here procedures for manual integration should be followed and records, including the authorization and justification for manual integration, should be maintained. The guidance fails to define what is manual integration: manual positioning of the baselines by an analyst (7,10,11).
From the wording in the guidance it appears that authorization is required each time that manual integration is needed. This is too draconian and manual integration must be justified in the validation report and the analytical procedure. Manual integration can occur daily if a method is analyzing biologicals, contrast media, or impurities. Scientific justification not regulatory diktat must be applied for manual integration. The PDA TR80 guidance (10) is the best guidance for integration as it has figures illustrating both good and bad integration practices, coupled with the differentiation between manual intervention and manual integration (11,12).
Data Management
Most of the section covers managing the administration of the computerized chromatographic data capturing and processing system (singular, this time!) better known as a CDS. The guidance does not say who is responsible but ideally this should be an IT function. True to form, there is another clause out of place with 14.2 requiring that data should be timely processed and reviewed, posing the question of why is this clause here and not under peak integration? If compliance with ALCOA+ requirements is required, why is there not a separate section for second person review, as this is where mistakes and errors should be caught in the laboratory. Clause 14.5 notes that printed records may be retained. May? May?? What planet are these writers on??? Strike three and out for quality and technical oversight and cue an extended bout of lalochezia.
References Section
There are three WHO references listed at the end of the document but these are only referenced to clause 14.1, which is most unhelpful. References to the United States and European Pharmacopoeias are equally unhelpful especially the specific chromatography and instrument qualification general chapters are not mentioned explicitly, for example, USP <621> (13), USP <1058> (14) and EP <2.2.46> (15).
Summary
The question posed by the title of these two columns is, “What is good about the WHO Good Chromatography Practices guidance?” Very little is my answer, and the document fails its SST criteria (if it had any). From the organization of the document, clauses in the wrong place, missing topics, wrong approaches to computerized system validation (CSV), to the inability to call a chromatography data system a CDS, it is a document that has been cobbled together without knowledge of the subject coupled with a total lack of technical and quality oversight. I’m not sure who this is written for and by whom.
This is a dangerous document if you were relying on it as your sole guide for good chromatography practices in a regulated laboratory. The writing is just too vague, generic and imprecise to be of real value: there is just one must and over 120 should statements in the document.
There is a need for a guidance document for Good Chromatographic Practice. However, with the problems and omissions presented here, the WHO guidance is not it. The current guide needs to be extensively rewritten, reorganized and expanded, ideally to highlight best practices that a laboratory should aim towards, if they do not work this way now.
Acknowledgements
I would like to thank Chris Burgess and Paul Smith for helpful review comments on the two parts of this column.
References
1) WHO Technical Report Series No.1025, Annex 4 Good Chromatography Practices (World Health Organization, Geneva, 2020).
2) R.D. McDowall, LCGC Eur. 33(11), 579–584 (2020).
3) T. Schofield, et al., “Distinguishing the Analytical Method from the Analytical Procedure to Support the USP Analytical Procedure Life Cycle Paradigm”, Pharmacopeial Forum, 45(6) (2019). Online only.
4) WHO Technical Report Series No.929, WHO Guidelines for Sampling of Pharmaceutical Products and Related Materials (World Health Organization, Geneva, 2005).
5) US Food and Drug Administration, Guidance for Industry: Out of Specification Results (FDA, Rockville, Maryland, USA, 2006).
6) R.D. McDowall, LCGC N. Am. 38(7), 411–419 (2020).
7) H. Longden and R.D. McDowall, LCGC Eur. 21(12), 641–651 (2019).
8) US Food and Drug Administration, Questions and Answers on Current Good Manufacturing Practices—Laboratory Controls (FDA, USA, 2019). Available from: https://www.fda.gov/drugs/ guidances-drugs/questions-and-answers-current-good-manufacturing-practices-laboratory-controls.
9) US Food and Drug Administration, 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products (FDA, Silver Spring, Maryland, USA, 2008).
10) Parenteral Drug Association, Technical Report 80: Data Integrity Management System for Pharmaceutical Laboratories (PDA, Bethesda, Maryland, USA, 2018).
11) R.D. McDowall, LCGC Eur. 28(6), 336–342 (2015).
12) R.D. McDowall, Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements (Royal Society of Chemistry, Cambridge, UK, 2nd ed., 2017).
13) United States Pharmacopoeia General Chapter <621> “Chromatography” (United States Pharmacopoeia Convention, Rockville, Maryland, USA).
14) United States Pharmacopoeia General Chapter <1058> “Analytical Instrument Qualification” (United States Pharmacopoeia Convention, Rockville, Maryland, USA).
15) General Text 2.2.46 “Chromatographic Separation Techniques,” European Pharmacopoeia (Strasbourg, France, 2005), pp. 69–73.
“Questions of Quality” Column Editor Bob McDowall is Director of R.D. McDowall Limited, Bromley, Kent, UK. He is also a member of LCGC Europe’s editorial advisory board. Direct correspondence about this column to the Editor‐in‐Chief, Alasdair Matheson: [email protected]
Articles in this issue
over 5 years ago
Modern Trends in Mixed-Mode Liquid Chromatography Columnsover 5 years ago
Flying High with Sensitivity and Selectivity:- GC–MS to GC–MS/MSover 5 years ago
A Review of MOSH and MOAH Analysis in FoodAdvertisement
Related Content
Advertisement



Advertisement
Advertisement