

Special Issues
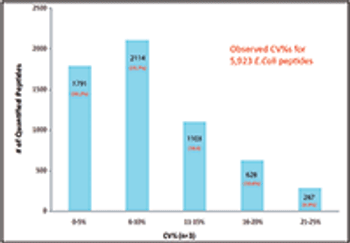
This article describes the development of a new data-independent acquisition (DIA) workflow for protein quantification that uses a mass spectrometer that combines three types of mass analyzers to achieve lower limits of detection (LOD), higher sensitivity, more accurate quantitative results, wider dynamic range, and better reproducibility than existing high-resolution accurate-mass (HRAM) tandem mass spectrometry (MS-MS) DIA workflows.




