Testing the Fundamentals: An Examination of Your Extraction Knowledge

As I write this, I’m also grading the final exam from my “Chromatography and Separations” course. We spent approximately the first 75% of the course covering column chromatography, capillary electrophoresis (CE), and field-flow fractionation. The remainder of the course focused on chromatographic sample preparation. The final exam was based on this last section of the course. Some questions make a good review of the fundamentals of various extraction techniques. The basis for these questions may have been discussed in previous iterations of this column, in analytical chemistry textbooks, or in books on chemical separations. I generally give the final exam in my graduate courses as a take-home exam, so you are free to use whatever resources you have available to you. Here’s your opportunity to test yourself, as the answers are provided.
Question 1: Consider a system comprised of water and a neutral solute. An aliquot of this mixture is used to partially fill a septum-capped vial, and solid-phase microextraction (SPME) is used to extract the solute from the headspace vapor. Using Gibbs’ Phase Rule, how many degrees of freedom describes this extraction system? Considering these degrees of freedom, what are the important parameters in developing this extraction?
The phase rule, F = C – P + 2, describes systems at thermodynamic equilibrium, where F is the number of degrees of freedom describing the system; C is the number of components in the system; and P is the number of phases. Because the phase rule applies to “pressure/volume/temperature (pvT)” systems, the constant 2 accounts for the pressure and temperature of the system. For example, if we have a single solute that may partition between an aqueous and an organic phase, there are three components (the aqueous and organic phases and the solute) and two phases (the aqueous and organic), so F = 3 – 2 + 2 = 3. Thus, if the temperature and pressure are constant, the concentration of solute in one phase dictates the concentration in the other phase because the system is at equilibrium. In other words, concentration is the third degree of freedom, which provides the basis for the concept of distribution ratios. In a slightly more complex, illustrative example, consider that the solute in the previous example is an amine. Now we have four components (the aqueous and organic phases, the deprotonated amine, and its protonated counterpart) with the two phases, or F = 4 – 2 + 2 = 4. As in the previous example, temperature, pressure, and concentration are degrees of freedom that describe the system. The fourth degree of freedom is the pH (relative to the pKb of the amine), which determines whether the amine is protonated or deprotonated.
Getting back to our question, in this case, we have four components (water, solute, headspace vapor, and SPME sorbent) and three phases (water, headspace vapor, and SPME sorbent), or F = 4 – 3 + 2 = 3. In addition to the temperature and pressure, because all three phases are in equilibrium with each other, [solute]aq ↔ [solute]g ↔ [solute]SPME, the concentration of solute in one phase dictates the solute concentration in all phases. Thus, headspace SPME can be considered a quantitative technique, provided the SPME fiber is exposed to the headspace for a sufficient period to allow equilibria to be attained.
Question 2: Refer to the graph in Figure 1 for a given pair of extractions. For each of the two data sets depicted, state what property limits the extraction from producing quantitative yields. How might you adjust the extraction conditions to produce a more quantitative yield in each case?


FIGURE 1: Extraction kinetics for two extraction examples, showing cumulative extraction yield versus the progress of the extraction.
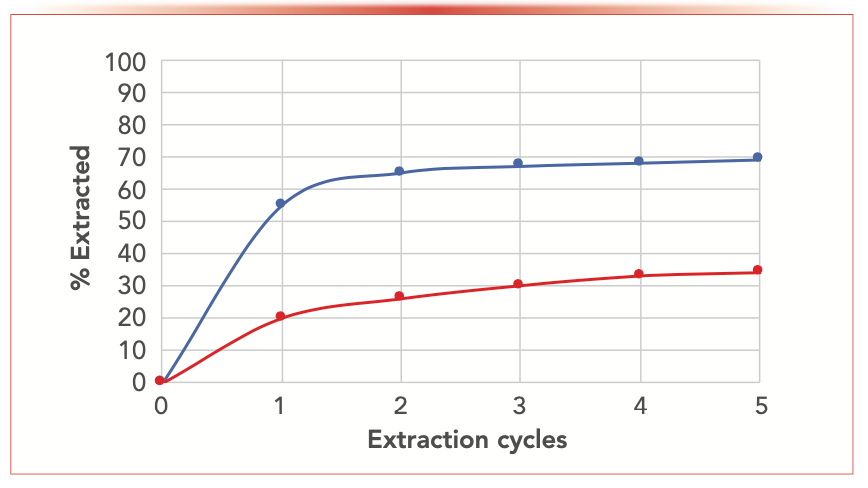
The kinetics of most extractions generally follows the curve shown in Figure 1. In these plots, the cumulative amount extracted is shown as a function extraction progress, which may be, for example, solvent volume or time in a flow-based extraction like pressurized liquid extraction (PLE) or supercritical fluid extraction (SFE), individual separative funnel extractions, or cycles in a Soxhlet apparatus. The kinetics show a rapid extraction period because of analyte solubility in the extraction solvent under the prescribed conditions, followed by a slowing of the curve, where the extraction kinetics become dependent on the diffusion of the solvent (with or without dissolved analyte) through the sample phase, asymptotic to the maximum amount extracted. Figure 1 demonstrates two different extraction examples.
In the upper blue curve, the initial slope is quite large, demonstrating high solubility of the analyte in the extracting solvent. Approximately 55% of the analyte is extracted in one cycle and a combined two-thirds following the second cycle. However, the total extraction never exceeds 70%. We can conclude that the rate-limiting step for this extraction is the diffusion of the extracting solvent into the pores of the sample particles (these diffusion-limited situations are much less prevalent when extracting liquid or gaseous samples) to solvate the analyte trapped inside the sample. There are two primary means of overcoming the diffusion limitation. The first is to shorten the diffusion pathlength (that is, increase the sample surface area) by grinding the sample. Extractions of solids are generally faster as the sample particle size decreases. For example, consider making a pot of coffee with whole beans as opposed to grinding the beans! The one thing to watch out for in this situation is to minimize grinding the sample too fine (say the consistency of flour or talcum powder) to avoid the particles packing together and impede solvent flow. The second means to address this situation is to enhance the diffusivity of the solvent. One may choose a less viscous solvent, but solubility is not the issue, so we would likely stay with the original solvent used. Supercritical fluids are one type of potential extracting solvent exhibiting fast diffusion rates. The easiest way to improve solvent diffusivity is to heat the system. As the temperature increases, the diffusion is faster, which is the principle behind techniques such as PLE or microwave-assisted extraction (MAE). PLE and MAE allow for higher temperatures than the atmospheric boiling point to be used. One caveat is that we must avoid thermal decomposition of the sample or analyte. Another less often used and more difficult way to increase solvent diffusivity is to add a secondary chemical, such as a surfactant or a gas like carbon dioxide (perhaps in the supercritical region), which lowers the surface tension and enhances diffusion.
The example shown in the lower, red curve in Figure 1 is limited by the analyte solubility in the extracting solvent. Only approximately 20% of the analyte is extracted during the first cycle before the curve flattens, though it still rises at a visible rate. This extraction is solubility limited. Again, increasing temperature generally results in increased analyte solubility. A different solvent, or a solvent additive, is most likely needed.
Plotting the extraction kinetics during method development, especially when extracting solid samples, often helps guide the extraction process: Should I switch solvents, heat, or grind the sample? However, the analyst should keep in mind two common situations where the shape of the kinetic plot may differ from that shown in Figure 1. The first common situation is where the analyte may decompose (because of light, heat, or oxidation) during the course of the extraction. The second common situation the analyte may be tightly adsorbed onto the sample matrix, or may even be encapsulated by the matrix. These situations may require more complex method development.
Question 3: Over the past 20–25 years, there have been serious concerns over the environmental consequences (for example, flammability, toxicity, and waste generation) of the use of organic solvents in chemical extractions. Discuss how modern extraction methods reduce or eliminate the use of organic solvents from the perspectives of (a) extracting liquid samples and (b) extracting solid samples.
Since the 1990s, the chemistry community has seen the rise of green chemistry, a thought process on the practice of our discipline considering the health and environmental consequences of our products and processes. At approximately the same time, there has been the concurrent development of the next generation of extraction techniques with improved performance. Techniques like solid-phase extraction (SPE), SPME, hollow-fiber membranes, SFE, PLE, and MAE are among the techniques that possess significant performance advantages compared to their traditional counterparts and happen to have green advantages as well. These approaches are preferred because methods cannot possess green advantages only; they must also be cheaper, faster, and more quantitative (equal performance will not cut it) than the methods they are replacing.
Extracting liquid samples has been greatly influenced by the use of adsorbents, such as chromatographic stationary phases. This led to the development of SPE, followed by SPME, stir-bar sorptive extraction (SBSE), immobilized liquid extraction (ILE), and others. The key feature of each of these techniques is that the dissolved (typically aqueous) sample is added to the sorbent phase, with a variety of configurations that define the techniques, and small amounts of organic solvent are used in the washing steps, the desorption steps, or both. The same organic solvents may be used in conventional extractions or different solvents may be used. As these methods, especially SPME and SBSE, were developed, thermal desorption of the analyte from the sorbent led to the complete elimination of organic solvent use. The various formats of these sorbent-based extractions became the basis for other techniques like hollow-fiber membranes and support-assisted liquid–liquid extraction (SALLE), which also dramatically reduced organic solvent use. Single-drop microextraction (SDME) can be envisioned as a derivative of SPME where microliters of immiscible organic solvent may be considered as the sorbent phase.
Meanwhile, the extraction of solid samples was advanced by the development of instrumental approaches that utilize energy to impact the extraction. Generally, the energy is manifested in the form of heat to improve solubility, diffusion, and desorption energy, as discussed in Question 2. These techniques include PLE, MAE, and ultrasound-assisted extraction (UAE). The advantage of these techniques is a reduction of solvent use from hundreds of milliliters in Soxhlet extraction, for example, to tens of milliliters. The techniques also happen to be faster (from up to a day to 15–30 min per extraction), and more reproducible.
In addition to the newer techniques described above, another approach applied to both liquid and solid samples is the use of alternative solvents with green advantages. Beginning in the mid-1980s, SFE began to be used more frequently. Supercritical carbon dioxide has solubility and selectivity advantages for certain compounds and favorable diffusion. The instrumental configuration of SFE also inspired the subsequent development of PLE and MAE. Natural products like D-limonene, α-pinene, ethyl lactate, deep eutectic solvents, and ionic liquids are all finding use in analytical extractions. Coacervative extractions, such as cloudpoint extraction (CPE), use surfactants to encapsulate solutes in the form of miscelles to isolate and separate the solutes from the liquid solution.
Question 4: Choose one of the more “advanced” methods (that is, automated Soxlet, UAE, PLE, SFE, or MAE) for extracting solid samples. Compare this method with the traditional Soxhlet procedure in terms of method operation, time, solvent use, cost, safety, and other performance variables.
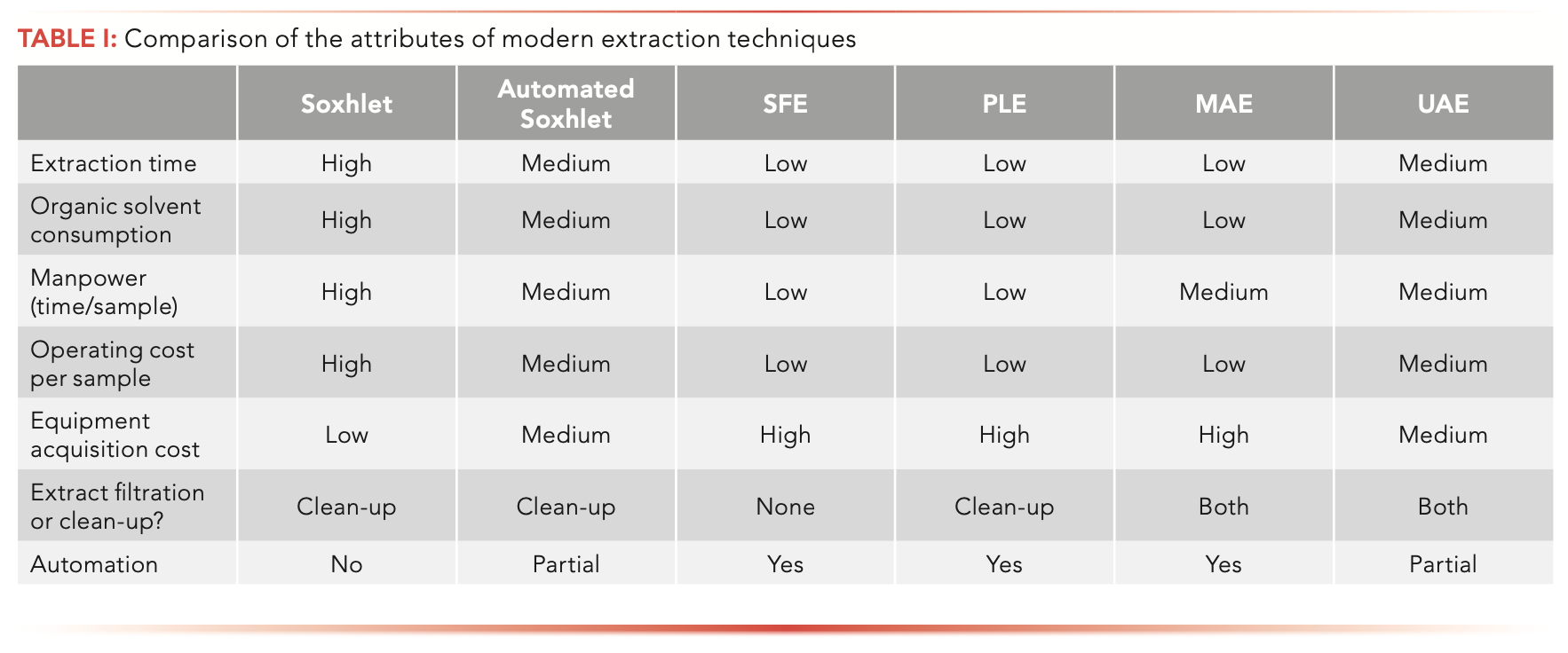
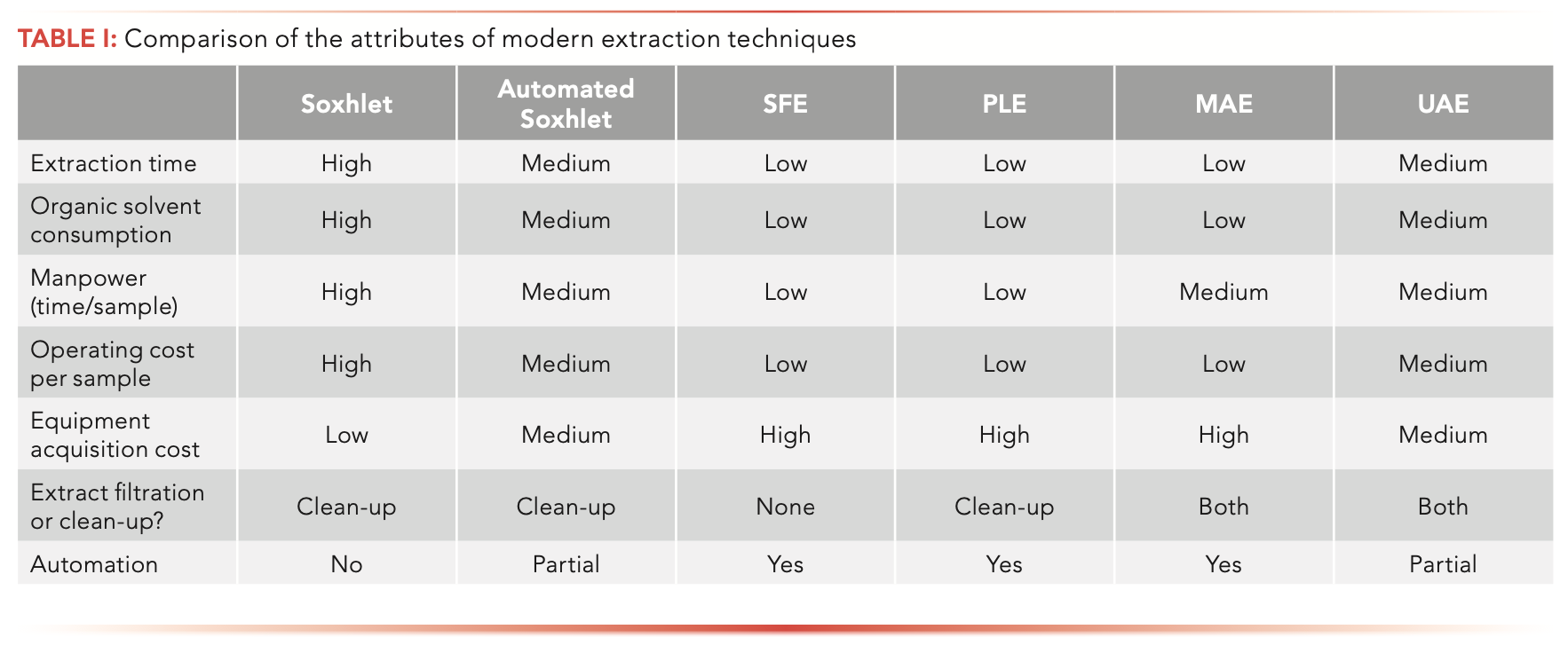
Table I summarizes the attributes of each of the subject extraction techniques. Each of the techniques is much quicker than Soxhlet extraction, which takes 6–24 h. SFE, PLE, and MAE use the least amount of solvent, even though automated Soxhlet and UAE are also reduced-solvent use techniques. The automation characteristics of SFE, PLE, and MAE result in less labor intensiveness. The instrumental methods suffer from a high acquisition cost, but that is amortized to a lower operating cost per sample. Although each technique is safe if operated properly (Soxhlet extraction should be performed in a fumehood), the instrumental methods have built-in features to avoid exposure to solvent vapors.

Question 5: Caffeine has an octanol–water partition coefficient, or Kow, of 0.616. A cup (237 mL) of coffee contains 95 mg of caffeine. If you want to extract caffeine from a cup of coffee using liquid–liquid extraction (LLE) with a separatory funnel and ethyl acetate as the solvent, how many extractions and how much solvent will be used to extract 92 mg of caffeine, if 237 mL each of coffee and ethyl acetate are used in each extraction?
The Kow can be used to estimate the amount of solvent used in a LLE. This value is tabulated for many common compounds of biochemical or environmental interest in several references. The Kow is the equilibrium constant showing the distribution of an analyte (A) between a nonpolar solvent (n-octanol) and water: Kow = [A]org/[A]aq, assuming n-octanol approximates all nonpolar organics. One thing I try teaching students is to estimate an answer using what is known. With a Kow of approximately 0.5, one-third of the solute should partition into the organic solvent with each extraction, or seven steps should extract approximately 95% of the caffeine in this case.
The equation:

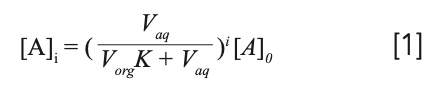
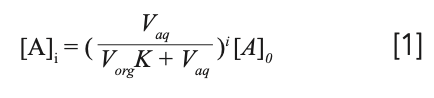
where [A]i is the concentration of analyte remaining in the sample following i number of extractions, [A]0 is the initial concentration of analyte in the sample, and Vaq and Vorg are the volumes of aqueous sample and organic extracting solvent, tells use the efficiency of an extraction. Using our ballpark estimate of seven extractions: [A]i = (237 mL coffee)/((237 mL ethyl acetate x 0.616) + 237 mL coffee))7 x 95 mg caffeine = 3.3 mg caffeine remaining. This will result in a whopping 1659 mL of ethyl acetate to extract the cup (237 mL) of coffee.
Therefore, seven extractions will result in a cup of coffee with 91.7 mg caffeine remaining. From an analytical perspective, selection of a solvent with a greater Kow for caffeine will provide a more efficient extraction. From a coffee drinker perspective, start with decaffeinated beans!
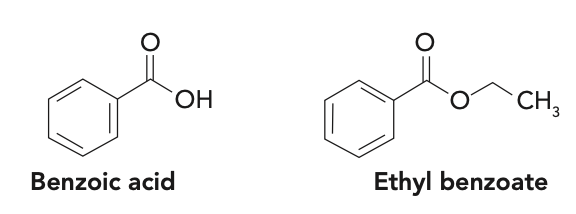
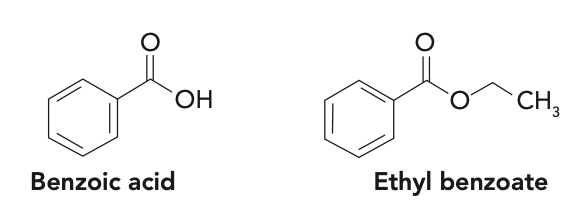
Question 6: Both benzoic acid and ethyl benzoate (structures shown below) are soluble in organic solvents. How can you use extraction to separate them from a solution?
Knowledge of the acid-base properties of benzoic acid is key to solving this problem, similar to the amine example in Question 1. The pKa of benzoic acid is 4.20, meaning that it will be in the deprotonated, or anionic, form at neutral pH or at any pH greater than 1.5 pH units higher than the pKa, or pH 5.60. Therefore, it should be water soluble. The ethyl ester is not impacted by pH and is essentially insoluble in water. Thus, a LLE using neutral water and a water-immiscible organic solvent should achieve the desired separation. As seen in Question 5, selection of the organic solvent should be based on the Kow of ethyl benzoate in that solvent.

Conclusion
I hope that this month’s column provides an understanding of important extraction and sample preparation concepts. You are challenged to recall your knowledge, or learn it for the first time, using the format of exam questions. Because our course is taught in alternate years, I’m likely to teach the course no more than one more time before retirement. Therefore, other professors are welcome to reuse or modify my questions, but it is also likely that these questions will be available to your students on various internet sites. Future installments of this column may present other exam questions I’ve developed over the years. In the meantime, as sung by Alice Cooper, “School’s out!”
ABOUT THE COLUMN EDITOR


Douglas E. Raynie
“Sample Prep Perspectives” editor Douglas E. Raynie is a Department Head and Associate Professor at South Dakota State University. His research interests include green chemistry, alternative solvents, sample preparation, high-resolution chromatography, and bioprocessing in supercritical fluids. He earned his PhD in 1990 at Brigham Young University under the direction of Milton L. Lee. Raynie is a member of LCGC’s editorial advisory board. Direct correspondence about this column via e-mail to LCGCedit@mjhlifesciences.com















Gulf Coast Conference: Increasing Density and Viscosity Throughput with Difficult Samples
October 19th 2023Daniel Wolbrecht, senior technical sales consultant at Anton Paar, held a workshop at the Gulf Coast Conference in Galveston, Texas, focusing on how heated autosampler units can help analyze difficult samples.


