Special Issues
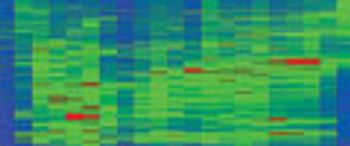
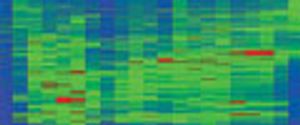
Because it is extremely rapid, biomarker discovery and identification using liquid chromatography–mass spectrometry (LC-MS), including both ion-trap and triple-quadrupole LC–MS, is well established. Fractionation of complex samples before LC–MS-MS analysis might be necessary to identify the proteins, greatly increasing the number of analyses required. In this case, there is ongoing debate regarding knowing whether the protein is identified correctly, knowing how much prior fractionation is needed to reduce complexity to the point where low-abundance proteins can be detected reliably, and balancing specificity with sensitivity.