The LCGC Blog: Charged Stationary Phases in Reversed Phase HPLC
We seem to have entered something of a mixed-mode world in reversed phase high-performance liquid chromatography (HPLC). Many stationary phases are now available that incorporate permanent or induced charges within, or alongside, hydrophobic alkyl-silica type bonded ligands.
Whilst the retention characteristic of these phases adds a dimension of complexity when planning, developing, or troubleshooting separations, the extra capabilities that these phases offer within the reversed phase separation space can be highly beneficial.
There are important distinctions to be made when considering phases in which the charge can be manipulated using pH adjustment of the eluent and those which are permanently charged, regardless of eluent pH. Stationary phases where the degree of surface charge can be manipulated using eluent pH, offer greater tunability of selectivity, but are more complex in terms of the requirements for accurate pH control, column (re)equilibration, and method development.
Phases which are permanently charged can feature the charged moiety within the alkyl chain, either closer to or further away from the surface, or by direct application to the silica surface.
In recent years, a category of stationary phases colloquially known as charged surface phases have been introduced to offer several chromatographic advantages. These phases have a balanced charge, which is sufficient to overcome several important chromatographic issues, especially when using mass spectrometry (MS) detection. These phases are designed to undergo lower energy interactions with ionogenic analytes when compared to mixed-mode phases, in which higher energy ion-exchange interactions are used to significantly alter analyte retention and selectivity.


Figure 1: Depiction of approaches to achieving a charged surface type C18 stationary phase.
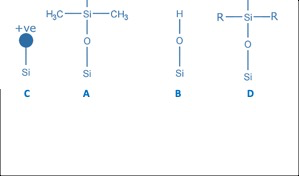
Figure 1A and B show a bonded C18 alkyl ligand alongside an uncharged lone silanol group, which remains post-ligand bonding and can undergo dipole interactions with polar analytes. When the pH of the eluent is raised above around pH 6–7, these silanol groups can ionise, and the resulting anionic surface can undergo significant secondary interaction with charged basic analytes, often leading to significant peak shape deformation. To combat this charge, surface columns are modified with a positive charge, which helps to shield ionogenic basic analytes from the silica surface, improving both peak shape and loadability when using low-ionic-strength eluents. The mechanism of achieving the surface charge is proprietary for all manufacturers, but potential methods include the use of a bonded, ionised, end-capping reagent, as shown in Figure 3D, and the direct modification of the silica surface by crosslinking with a reagent which includes an ionogenic moiety, as shown in Figure 3A.
These charged surface phases offer several advantages to the chromatographer, including:
- Alternative selectivity
- Improved peak shape and loading for basic analytes in low ionic strength mobile phases
- Reduced re-equilibration times when switching from high to low pH columns.
Of the listed advantages, perhaps the most important are peak shape and loadability improvements, driven mainly by modern approaches to eluent design when using MS detection.
To maximise sensitivity with MS detection, chromatographers are choosing to reduce the ionic strength of eluent systems or simply to alter pH using weaker acids or bases and omit the use of a salt counterion. Whilst this approach may leave the analysis more susceptible to changes in retention time and selectivity, this is often outweighed by the improvements in detector sensitivity through the lowering of charge competition within the sprayed droplets in electrospray atmospheric pressure ionisation.
Poor peak shape, especially for charged basic analytes, is a well-documented phenomenon and is caused by several factors, especially in low-ionic-strength eluent systems (1-3). The interaction of the cationic analyte with surface anionic sites, predominantly charged surface silanol groups, can cause secondary interactions, which result in peak tailing. While the use of high-purity silica and advanced silica bonding and deactivation (end-capping) regimes have reduced these phenomena, it can remain problematic. The loadability of stationary phases for charged analytes in low-ionic-strength eluents can be orders of magnitude less than neutral analytes. Additionally, peak tailing can occur even with very low loadings of charged analytes, depending upon the ionic strength of the eluent, the surface area of the packing material, extent of analyte ionisation and the use and concentration of a counter ion (ion-par), such as trifluoroacetic acid.
The use of an intentionally introduced, ’ow-level, positive charge on the silica surface can overcome secondary interactions with surface silanol groups, essentially by providing a repulsion effect toward cationic analytes, i.e. a basic analyte at eluent pH significantly below the pKa value. The mechanism for increased loadability of the charged surface phases is not well understood but can be demonstrated empirically through the application of varying degrees of surface charge. With too little surface charge, peaks for charged basic analytes at low-eluent pH tend to suffer from bi-Langmurial, or fronting, peak behaviour, and with too great a surface charge, they suffer from anti-Langmurial, or tailing behaviour, suggesting an optimum surface charge is required to produce Gaussian, neither fronting nor tailing, peak shapes. When analysing charged basic analytes at low concentrations, peak shape is important to take advantage of other improvements in column efficiency, such as reduced dimension particles.
Whilst the extent and strength of ion-exchange behaviour in these charged surface phases is intended to be lower than that used in more traditional mixed-mode type phases, there can be an impact on the selectivity of the stationary phase. This depends upon the degree of surface charge, the nature of the bonded phase ligand (and therefore the accessibility of the charged silica surface), the extent of ionisation of the analyte molecules, and their charge state (cationic or anionic). It is important to highlight that while a cationic surface charge is convenient in the repulsion of basic analytes, and therefore their improved peak shape, these charges may also act to increase retention for any charged acidic analytes present in the sample.
To further highlight this point, a comparison was made between various columns of nominal L1 classification (C18) to investigate the relative retention of acidic, neutral, and basic species on traditional C18 and charged surface C18 phases. We used a recently updated version of the Hydrophobic Subtraction Model (HSM2) (4) to compare relative retention of acidic and basic analyte test probes using calculated (simulated) chromatograms. The model uses ethyl-benzene retention time as a comparator for each test probe and uses these relative retention times to describe various interactions between analyte and stationary phase including: hydrophobic retention, hydrogen bonding, cation exchange, accessible volume and dipole interaction according to Equation 1;

- solute / column hydrophobicity
b (A) – solute / column hydrogen bond basicity
a0 (B) – solute / column hydrogen bond acidity
k © – solute / column cation exchange ability (protonated bases at pH 2.8)
v (V) – solute / column accessible volume (analyte hydrodynamic volume)
d (D) – solute / column dipole parameter")
h (H)- solute / column hydrophobicity
b (A) – solute / column hydrogen bond basicity
a0 (B) – solute / column hydrogen bond acidity
k © – solute / column cation exchange ability (protonated bases at pH 2.8)
v (V) – solute / column accessible volume (analyte hydrodynamic volume)
d (D) – solute / column dipole parameter
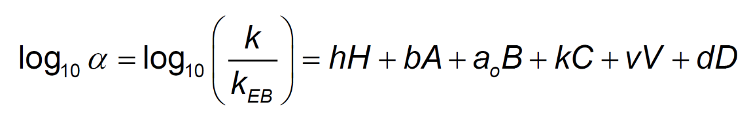
The columns chosen for the comparison were from the same manufacturer with the same stationary phase particle size, the same underlying silica, and the same bonded phase ligand (C18), ensuring that any differences in analyte retention were predominantly due to the applied surface charge.
Column parameters (H, A, B etc.) for each of the analyte/stationary phase interaction descriptors can be made using empirical measurements of a variety of test probes with carefully chosen physicochemical properties. The values of these column parameters for the chosen test columns are shown in Figure 2.


Figure 2: Relative magnitude of each analyte/stationary phase interaction terms within the revised Hydrophobic Subtraction Model for a C18 bonded column and a charged surface variant of the same column.

It is interesting to note, among other differences between the phases, that the cation exchange capacity (C Term) of the charged surface stationary phase is considerably lower, in keeping with the theory that the positively charged surface shields basic analytes from interaction with any anionic residual silanol groups on the silica surface. Further, the hydrogen bond acidity (A Term) of the phase describes the ability of non-ionised silanol residues to interact with non-ionised basic analytes. Again, the lower value of this parameter is consistent with the shielding (repulsive) effect of the positive surface charge preventing these interactions.
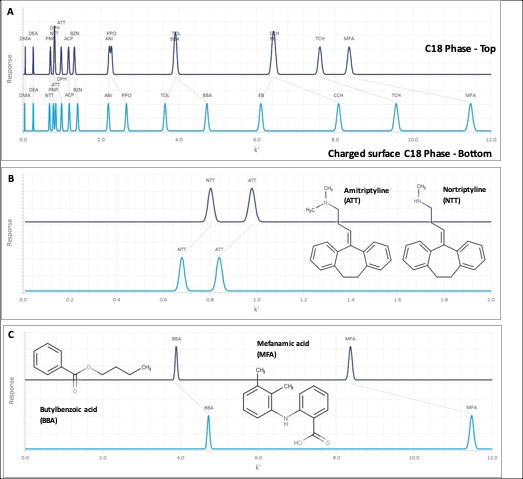
In the absence of the original screening retention times for the columns within the database, it is possible to use the hydrophobic subtraction model (HSM) equation values for analytes and stationary phases to reconstruct a simulated chromatogram of the 16 test probes of the HSM. These simulated chromatograms are shown in Figure 3.

 and C18 charged surface (lower chromatogram) Columns derived from the revised Hydrophobic Subtraction Model. A: Full Chromatogram for all 16 test probes, B: Charged Basic Analytes, C: Charged Acidic Analytes.")
Figure 3: Simulated chromatograms for C18 (top chromatogram) and C18 charged surface (lower chromatogram) Columns derived from the revised Hydrophobic Subtraction Model. A: Full Chromatogram for all 16 test probes, B: Charged Basic Analytes, C: Charged Acidic Analytes.
The relative retention factors for the acidic and basic analytes highlighted in Figure 3 are shown in Table 1, alongside the relative magnitude in the retention factor change.


Table 1: Retention Factors for analytes in simulated chromatograms from Figure 3 and % change for each analyte between C18 and C18 charged surface phases.

It is interesting to note that the relative changes in retention behaviour fit well with the theoretical behaviour of charged surface columns, i.e. a decrease in the retention of basic species due to the slight repulsive effects of the cationic surface charge and an increase in the retention of acidic analytes due to an ion exchange interaction with the cationic surface charges.
It should be noted that in deriving the HSM2 parameters, the initial test probe retention time measurements were undertaken at approximately neutral pH with a 60 mM phosphate buffer. Whilst the acidic and basic analytes shown in Figure 3 will have been fully ionised at the experimental pH, the effects of the higher buffer concentration on analyte/stationary phase interactions on the charged surface cannot be predicted. We have compared several C18/charge surface C18 columns pairs using this approach, and all have shown the same behaviour, with a slight decrease in retention of charged basic compounds and an increase in retention of charged acidic compounds.
It is clear from Figure 3 (Top) that the simulated test probe chromatograms for the C18 and the charged surface equivalent show significant selectivity differences, and this should be borne in mind by chromatographers wishing to take advantage of the improved peak shape of charged basic analytes in low ionic strength mobile phases.
In terms of improved peak shape of basic analytes, much is demonstrated in column manufacturer literature to show that this claimed advantage is born out in practice and is therefore an advantage for chromatographers wishing to analyse charged basic analytes in low ionic strength mobile phases and to attain the maximum sensitivity.
References
(1) McDonald, P.D., Bidlingmeyer, B. Preparative Liquid Chromatography; Elsevier, 1986.
(2) Snyder, L. Principles of Adsorption Chromatography, Elsevier, Marcel Dekker, 1967.
(3) Guichon, G., Shirazi, S.G., Katti, A., Fundamentals of Preparative and Non-Linear Chromatography; Academic Press, 1994.
(4) Stoll, D.R., Dahlseid, T.A., Rutan, S.C., et al. Improvements in the predictive accuracy of the hydrophobic subtraction model of reversed-phase selectivity, J. of Chromatogr. A. 2021, DOI: 10.1016/j.chroma.2020.461682









TD-GC–MS and IDMS Sample Prep for CRM to Quantify Decabromodiphenyl Ether in Polystyrene Matrix
April 26th 2024At issue in this study was the certified value of decabromodiphenyl ether (BDE 209) in a polystyrene matrix CRM relative to its regulated value in the EU Restriction of Hazardous Substances Directive.

LC–MS/MS-Based System Used to Profile Ceramide Reactions to Diseases
April 26th 2024Scientists from the University of Córdoba in Córdoba, Spain recently used liquid chromatography–tandem mass spectrometry (LC–MS/MS) to comprehensively profile human ceramides to determine their reactions to diseases.


High-Throughput 4D TIMS Method Accelerates Lipidomics Analysis
April 25th 2024Ultrahigh-pressure liquid chromatography coupled to high-resolution mass spectrometry (UHPLC-HRMS) had been previously proposed for untargeted lipidomics analysis, but this updated approach was reported by the authors to reduce run time to 4 min.


