

|Articles|November 1, 2020
- LCGC Europe-11-01-2020
- Volume 33
- Issue 11
What’s Good About the WHO Good Chromatography Practices Guidance? Part 1
Author(s)R.D. McDowall

Listen
0:00 / 0:00
Key Takeaways
- The WHO's Good Chromatography Practices document is criticized for its disorganized structure and lack of specificity, failing to meet ALCOA+ criteria.
- Critical aspects like software validation, cybersecurity, and technical controls are inadequately addressed, undermining data integrity.
In September the World Health Organisation (WHO) issued a new guidance document on Good Chromatography Practices. What guidance does it contain and is it useful? Has the document failed its SST acceptance criteria?
Advertisement
Chromatography data systems (CDSs) have been at the heart of data integrity falsification, fraud and poor data management practices for 15 years since Able Laboratories 483 Observations were published by the FDA in 2005 (1,2). Although regulatory authorities have published much guidance on data integrity (3–7) there is little detailed guidance for chromatography data systems (CDS) except for the Parenteral Drug Association (PDA) Technical Report 80 where there is a good section on peak integration (8).
In September, the World Health Organisation (WHO) published Technical Report Series (TRS) 1025 which is the 54th report from the WHO Expert Committee on Specifications for Pharmaceutical Preparations, a slimline 345 pages for download. Tucked away in Annex 4 is a section entitled Good Chromatography Practices of obvious interest to the readership of LCGC Europe (9). As I’m reading the document and writing this column, I can attest to the effectiveness of lalochezia or using swearing to relieve frustration.
The Curate’s Egg
The WHO Good Chromatography Practices (GChromP) document is best described as a curate’s egg. This expression was coined from a cartoon published in a British satirical magazine in the late 19th century that pictured a curate (assistant to a vicar) having breakfast with a Bishop and the curate is eating a rotten egg. When the Bishop points this out the curate responds politely, if rather stupidly, that the egg is good in parts. The same can be said of the WHO chromatography guidance document (9). However, there are more bad parts than good which makes this a poor guidance document for chromatography.
Dopey Document
Anything issued by a regulatory authority should be exemplary to demonstrate how document controls should be implemented. This is not the case here. The numbering of the clauses within the document does not comply with ALCOA+ criteria as clause 1.4 is missing and there are two 6.1 clauses and two 8.1 clauses (9). How can you trust a guidance that can’t get the simplest things right? Anyone for accuracy? Quality oversight appears conspicuous by its absence.
What is the Aim of this Guide?
Here’s where the problems start. The “Introduction” and “Scope” section has five clauses (even though it finishes at 1.6) that make general statements about the use of chromatography in QC laboratories, the sample types that can be analysed and the types of the methods that can be used. Intermingled with this are statements:
- Observations during inspections have shown that there was a need to a specific good practices (GXP) document (clause 1.1)
- Owing to the criticality of results obtained through chromatography, it must be ensured that data acquired meet ALCOA+ principles … (clause 1.3)
- This document provides information on GXP to be considered in the analysis of samples when chromatographic methods and systems are used (clause 1.4) (9).
Here and throughout the document, there are very general and non-specific statements. Statements are often clarified with conditioning words and phrases such as “where appropriate”, “it is preferable”, “may”, “should” (there are 121 “shoulds” vs. one “must” in the document). There is no reference to the WHO Good Records Management guidance and Annex 1 containing the best description of the ALCOA criteria (5) except in clause 14.1 at the end of the document. A focused introduction with emphasis on good record keeping and data integrity practices would have been preferable.
Structure of the GChromP Guidance
The document structure is curious as sections appear to have been arranged as if pulled at random from a hat with one or two options left behind. The two main sections dealing with set up and system are sections 3 and 8. Now I fail to understand why section 8 on an electronic system that includes important information on system architecture (standalone versus network) is stuck in the middle of the document when it should be next to or integrated with section 3 that discusses supplier and system selection.
What is equally baffling is the total omission of the phrase chromatography data system from the entirety of the document. Instead readers are treated to a comedy of errors in calling a CDS an Electronic Data Management System (EDMS) (8.1) or computerised chromatographic data-capturing and processing system (13.2). What would have been much better is to have sections 3, 4, and 8 together with sections 5–7 on set up and configuration of the application software and interfacing of the chromatographs.
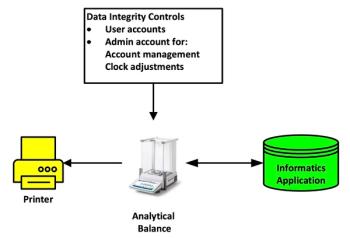
Look at the chromatography process, shown in green in Figure 1, in the words of that great analyst, Commander Spock, it is a chromatography process, but not as we know it. Presented in sections 9–14 is a selection of tasks but when seen objectively cannot be considered a chromatography process. Emmental anyone? After more lalochezia, I will continue.
Sins of Omission
Apart from the wacky order of sections, what is missing from this document? Here’s a list at the system level:
- A CDS with a database is preferable to one that stores data in operating system directories
- There is no discussion of how to avoid hybrid systems and the problem of linking paper printouts with the CDS electronic records (10)
- Avoid using spreadsheets as discussed in a recent Questions of Quality column (11) as all SST calculations are standard in CDS software and using custom calculations means that all records are in one location
- Cybersecurity to protect all chromatographic data is conspicuous by its absence
- Avoid using USB sticks to transfer data
- Leveraging supplier’s software development work to reduce the amount of in-house computer validation is not mentioned (12)
- There is no mention of using the CDS settings to protect e-records or use electronic signatures which are documented in a configuration specification
- There is no mention of the advantages of using the technical controls that are validated once and used multiple times versus inefficient, error prone and manual procedural controls as discussed in the WHO’s own data integrity guidance (5).
Goofy Glossary
Section 2 is the glossary with 12 definitions not defined in existing WHO sources (9) plus a cross reference to national pharmacopoeias. However, the terms for audit trail, backup, (computerised system) validation, data, data integrity and metadata have been published already in WHO TRS 1019, Annex 3, Appendix 5, a year earlier (13). There are three definitions that need to be discussed now:
- Sample set: This term should be sequence file as sample set is used in a commercial CDS application and should not be used in any guidance document. The same is true for system and project audit trails later in the document.
- Validation: This is defined as The action of proving and documenting that any process, procedure or method actually and consistently leads to expected results (9). This definition is wrong as it does not include software within its scope and does not mention the specification of the system to be validated. Moreover, there is a much better definition In WHO’s TRS 1019 where the definition of computerised system validation is Confirmation by examination and provision of objective and documented evidence that a computerised system’s predetermined specifications conform to user needs and intended use and that all requirements can be consistently fulfilled (13).
- Source data: This definition is wrong as it fails to understand the term. Source data or raw data has been copied from the MHRA’s GXP data integrity guidance (4) which is also wrong (14). Source data is defined as Original data obtained as the first-capture of information, whether recorded on paper or electronically (9). This is wrong. Source data is a Good Clinical Practice term and is defined in ICH E6(R2) 1.51 as All information in original records and certified copies of original records of clinical findings, observations, or other activities in a clinical trial necessary for the reconstruction and evaluation of the trial. Source data are contained in source documents (original records or certified copies) (15). This ICH definition is equivalent to GLP definitions of raw data in FDA and OECD regulations (16,17) and also EU GMP Chapter 4 raw data (18) and FDA GMP complete data in 21 CFR 211.194(a) (19). The scope of complete data and raw data have been described in earlier Questions of Quality (20) and Data Integrity Focus publications (21) and must consist of ALL data and records generated during analysis. Incidentally, a recent draft OECD GLP guidance on data integrity (22), based on MHRA guidance documents (3,4) now has a correct definition of raw data that is consistent with source data and GLP raw data definitions discussed above.
An omission from the glossary is a definition of dynamic and static data. This is necessary as chromatography data are dynamic as they must be interpreted with the requirement to retain them in dynamic format throughout the record retention period. This is stated in the WHO data integrity guidance issued in 2016 (5) as well as the MHRA (4) and FDA guidance documents (6) but there is no cross reference in the GChromP guidance. Please excuse me for a short break to shout some words from the Anglo-Saxon portion of the English language.
Chromatographic Systems and Electronic Systems
These are the titles of sections 3 and 8 respectively and there are two main questions.
- Why are they separated?
- Why is there no specific mention of chromatography data system software?
The former section presents clauses requiring the system to meet regulatory and GXP requirements (is there a difference?) and meet ALCOA+ principles. Summarising the requirements here, there need to be agreements outlining responsibilities between the supplier and the purchaser, supplier selection and qualification should ensure that hardware and software are suitable for intended use and suitably sited with the correct environmental conditions. However what hardware are we talking about: chromatographs, application servers, data storage, IT infrastructure or all of the above? This section appears to suggest that qualification by the supplier is acceptable but a supplier has no control over a laboratory’s intended use.
The problems mount when we consider section 8, strangely entitled electronic systems. No mention of CDS software at all but bizarrely the controlling application is now called an Electronic Data Management System (EDMS)! The consideration of standalone and networked systems is discussed here with the preference for the latter and the former to be risk assessed and appropriately managed, whatever that means. In the four-part series by Burgess and McDowall on the ideal chromatography data system, we stated that standalone systems are inadequate and inappropriate for a regulated laboratory (23–26). We make the point in the series that even for a single chromatograph a networked virtual server is possible to avoid storing data on a standalone PC. However, the major issue is why is this point not integrated with section 3 as this is a key consideration for selection of the CDS application? It should not be dumped in the middle of the document like an island in an ocean of stupidity.
Omitted from either section is the need for a database to organise chromatographic data rather than files stored in directories in the operating system where they are more vulnerable and audit trails are ineffective. Nowhere in the document is the need for documenting the configuration of the software to protect electronic records. The document also lacks a discussion of technical versus procedural controls and why technical controls are preferable, as discussed in several places in the WHO’s own guidance from 2016 (5), yet there is no mention of them here or a specific reference this guidance.
There is also the requirement in 13.2 that chromatographs should be interfaced to a CDS (rather than detector output being fed to a chart recorder). I appreciate that in some countries where WHO operates there are equipment limitations, but this statement would be much better placed in section 3 or 8 and not at the back of the document. Specification of a chromatograph and CDS software was discussed in a recent Questions of Quality column to give an idea of what is entailed in this process that should be done before selecting the supplier (27).
Qualification, Validation, Maintenance and Calibration
Although this section covers the four items in the heading I want to focus on the initially on validation of the software. Incidentally, software is not mentioned anywhere in this section which is a critical failing. The problem I have is the approach to the validation of a CDS does not match reality and I doubt the technical and practical expertise of the writers of this section as well as the quality oversight which is either woefully inadequate or non-existent.
Section 4.3 states that all stages of qualification (this should be validation) should be considered. But of what? The scope of the qualification is not stated. However, whatever we are qualifying we need a user requirements specification (URS), design qualification (DQ), factory acceptance tests (FAT), site acceptance tests (SAT), installation qualification (IQ), operational qualification (OQ) and performance qualification (PQ). This is stupid ,from a personal perspective of 35 years experience of validating laboratory informatics applications and qualification of analytical instruments I have never required FAT or SAT as this is applicable to production equipment as shown in my book on the subject (12). There is the grave omission of documenting the software configuration to protect the electronic records or implement electronic signatures and the need for user requirements to be traceable throughout the life cycle (12).
Interestingly, the reference for computerised system validation (CSV) is quoted as WHO Technical Report Series 1019 Annex 3, Appendix 6 (20), but the requirements for CSV are found in Annex 3, Appendix 5 (13), referenced in further reading! Pay attention! Are you following this? In Appendix 5 on CSV there is NO reference to FAT and SAT but CSV does require a configuration specification to record the application settings. However, Appendix 6 for Guidelines on Qualification which is mainly for production equipment has requirements for FAT and SAT (28). This is not a typographical error. Strike one for quality and technical oversight of this guidance.
Access and Privileges
Definition of user roles and their access privileges is presented in section 5 (9). This is relatively good, requiring a procedure for user account management, a list of current and historical users, no conflicts of interest with the allocation of administrator rights. Clause 5.5. notes that the settings on paper should match those allocated electronically (presumably in the CDS software that is not mentioned, again). I disagree with one item in 5.1 that the SOP for user account management covers the creation and deletion of user groups and users. There is no mention of modification which is typical in many laboratories. For example, a new hire will be assigned a trainee role and after a period of assessment their privileges will be modified to analyst. Also, I disagree with deletion of user groups and users; best practice is to disable an account as this leaves the user identity in the system and does not allow the creation of a new user with the old identity. This is an essential requirement for ensuring attribution of action to an individual i.e. the first A in ALCOA+.
Audit Trail
This section has a fatal flaw in clause 6.1 that renders the whole section useless by omitting monitoring deletions of data (9).The writers may hide behind for example but guidance needs to be explicit especially when it comes to audit trail functionality. In contrast, the definition of audit trail in the glossary does include deletion. Strike two for consistency and quality oversight.
In 6.3 the clause mentions system and project audit trails which with sample set is explicit terminology for the functions for a commercial CDS application. As such these should not be part of a non-partisan guidance document from a regulatory authority.
Date and Time Stamps
Date and time stamps are important for ensuring data integrity and it is good that there is a section on the subject. However, like the curate’s egg there are a few omissions. Section 7.2 requires the clock to be locked which is a bit stupid if the CDS is installed on a standalone PC with no option to correct time. A better phrasing would be to state that access to and changes of the clock should be restricted to authorised individuals and documented. For networked CDS applications, there is no mention of a trusted time source to ensure the time is correct within a minute as stated in a withdrawn draft FDA guidance on time stamps (2002). There is no mention that the time and date format should be unambiguous and documented in specifications for the system.
In addition, what happens with summer and winter time changes? The only guidance on this topic comes from an FDA Guidance for Industry on Computerised Systems in Clinical Investigations:
Controls should be established to ensure that the system’s date and time are correct. The ability to change the date or time should be limited to authorized personnel, and such personnel should be notified if a system date or time discrepancy is detected. Any changes to date or time should always be documented. We do not expect documentation of time changes that systems make automatically to adjust to daylight savings time conventions (29).
However, the configuration of the operating system to adjust time automatically should be included in the specification documents as well a statement that the change will not be recorded.
To Be Continued...
In the second part of this review of the WHO’s Good Chromatography Practices guidance document we will look at what the guidance says for performing chromatographic analysis.
References
- Able Laboratories Form 483 Observations. (2005). Available from: https://www.fda.gov/media/70711/download.
- R.D.McDowall, LCGC Europe 27(9),486–492 (2014).
- MHRA GMP Data Integrity Definitions and Guidance for Industry 2nd Edition, Medicines and Healthcare products Regulatory Agency: London (2015),.
- MHRA GXP Data Integrity Guidance and Definitions, Medicines and Healthcare products Regulatory Agency: London (2018)).
- WHO Technical Report Series No.996 Annex 5 Guidance on Good Data and Records Management Practices, World Health Organisation: Geneva (2016).
- FDA Guidance for Industry Data Integrity and Compliance With Drug CGMP Questions and Answers Food and Drug Administration: Silver Spring, MD (2018).
- PIC/S PI-041-3 Good Practices for Data Management and Integrity in Regulated GMP / GDP Environments Draft, Pharmaceutical Inspection Convention /Pharmaceutical Inspection Cooperation Scheme: Geneva (2018)..
- Technical Report 80: Data Integrity Management System for Pharmaceutical Laboratories, Parenteral Drug Association (PDA): Bethesda, MD (2018).
- WHO Technical Report Series No.1025, Annex 4 Good Chromatography Practices, World Health Organisation: Geneva. (2020)
- R.D.McDowall, LCGC N. America 38(2): 82–88 (2020)..
- R.D.McDowall, LCGC Europe 33(9) 468–476. (2020)
- R.D.McDowall, Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements Second Edition ed., Cambridge: Royal Society of Chemistry (2017).
- WHO Technical Report Series No.1019 Annex 3, Appendix 5 Validation of Computerised Systems. World Health Organisation: Geneva (2019).
- R.D. McDowall, Spectroscopy 33(12): 8–11 (2018).
- ICH E6 (R2) Guideline for Good Clinical Practice, International Conference on Harmonisation: Geneva (2018).
- 21 CFR 58 Good Laboratory Practice for Non-Clinical Laboratory Studies. Food and Drug Administration: Washington, DC (1978)
- OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring Number 1, OECD Principles on Good Laboratory Practice, Organsation for Economic Co-operation and Development: Paris (1998).
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 4 Documentation, E. Commission, Editor. Brussels (2011).
- 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products, Food and Drug Administration: Sliver Spring, MD (2008)..
- R.D.McDowall, Spectroscopy 31(11) 18–21 (2016).
- R.D.McDowall, LCGC N.America 37(4): 265–268 (2019)).
- OECD Draft Advisory Document of the Working Group on Good Laboratory Practice on GLP Data Integrity, Organisation for Economic Cooperation and Development: Geneva. (2020)
- R.D.McDowall and C.Burgess, LCGC N. America 33(8), 554–557 (2015).
- R.D.McDowall and C.Burgess, LCGC N. America 33(10), 782–785 (2015). .
- R.D.McDowall and C.Burgess, LCGC N. America 33(12), 914-–917. (2015.)
- R.D.McDowall and C.Burgess, LCGC N. America 34(2), 144 –149 (2016).
- R.D.McDowall, LCGC Europe 33(5), 257–263 (2020)..
- WHO Technical Report Series No.1019 Annex 3, Appendix 6 Guidelines on Qualification, World Heath Organisation: Geneva (2019),
- FDA Guidance for Industry Computerised Systems Used in Clinical Investigations, Food and Drug Administration Rockville, MD, (2007).
“Questions of Quality” editor Bob McDowall is Director of R.D. McDowall Limited, Bromley, Kent, UK. He is also a member of LCGC Europe’s editorial advisory board. Direct correspondence about this column to the editor‑in‑chief, Alasdair Matheson, [email protected]
Articles in this issue
Advertisement
Related Content
Advertisement



Advertisement
Advertisement
Trending on LCGC International
1
LC-MS/MS Links Vitamin D Levels to Sleep Timing
2
HPLC, PTR-ToF-MS Enable AI Grading of White Tea
3
How Should You Respond to FDA 483 Observations?
4
LC-MS/MS Finds PFAS Migration from Stuffed Toys
5