

|Articles|February 1, 2022
- February 2022
- Volume 35
- Issue 02
- Pages: 66–71
How Static Are Static Data?
Author(s)R.D. McDowall

Advertisement
A balance printout is a fixed record, and is also called static data. But how static are static data when the weight is used in a chromatographic analysis? Also, have some regulatory data integrity guidance documents failed to comply with their own regulations?
Data integrity continues to be a major topic for regulated pharmaceutical laboratories, which has resulted in health authorities issuing guidance documents to ensure the reliability and integrity of good manufacturing practice (GMP) records (1–5). Some of the guidance refers to GMP records and data as being either static or dynamic (2–5). Static and dynamic data are generally accepted terms, as seen in other data integrity publications (6–8).
Defining Static and Dynamic Data
Table 1 presents the definitions of static and dynamic data, with examples quoted from question 1(d) from the FDA Data Integrity Guidance (4). The definition of static data gives the impression that nothing happens after the record is created and they remains immutable for the record retention period. In contrast, dynamic data require interpretation by an analyst to obtain the reportable result. As you can see from Table 1, chromatography data and spreadsheets are examples of dynamic data; static data involve a balance printout.
What I want to discuss in this “Questions of Quality” instalment is that some static data are inputs into dynamic data processing. In these circumstances, static data are converted into dynamic data. However, creating a static record where the weight is manually entered into a chromatography data system (CDS) is an inefficient and error-prone process. Instead, laboratories should automate the complete process to eliminate the static data and enable sharing of all analytical data for review and to comply with the complete criterion of ALCOA+. Furthermore, the focus on printouts from analytical instruments (5) is inconsistent with US and EU regulations for keeping current with technology (9,10), in addition to some recent FDA warning letters (11–13) that will be discussed later.
Static and Dynamic Data in Practice
CDS dynamic data as presented in Table 1 require a user to interpret peak integration for each data file in the sequence. One more requirement for dynamic data is that they must remain dynamic throughout the record retention period (2), therefore:
- Electronic records are primary and printouts are secondary in the data set.
- Paper alone can never be the GMP record from an electronic record generated by a computerized system.
- Hybrid records are tolerated currently but two data integrity regulatory guidance documents encourage their replacement (3,5).
- Dynamic data cannot be printed to PDF and stored in an informatics application. The PDF file is a static electronic record as a user cannot interact with it.
In contrast, once static data have been created, the user cannot interact with them. Right?
But are static data really static with no further user interaction?
How Static are Static Data?
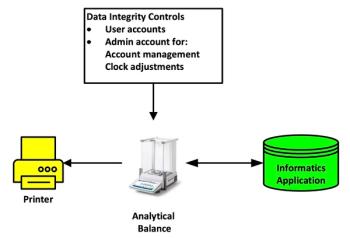
Back to the laboratory bench and imagine you are weighing either sample aliquots or a reference standard on an analytical balance and the weighing sequence (for example, vessel weight, tare, material weight) is printed out. It is generally accepted that this is a static record where the balance printout is attached to a laboratory notebook or analytical batch record. Indeed, the 2021 PIC/S guidance in section 8.9.1 (5) states:
Some very simple electronic systems, e.g. balances ….which do not store data, generate directly-printed paper records. These types of systems and records provide limited opportunity to influence the presentation of data by (re-)processing, changing of electronic date/time stamps. In these circumstances, the original record should be signed and dated by the person generating the record and information to ensure traceability, such as sample ID, batch number, etc. should be recorded on the record. These original records should be attached to batch processing or testing records.
This approach is repeated in the FDA data integrity guidance in question 10 (4):
10. Is it acceptable to retain paper printouts or static records instead of original electronic records from stand-alone computerized laboratory instruments, such as an FT-IR instrument?
A paper printout or static record may satisfy retention requirements if it is the original record or a true copy of the original record (see §§ 211.68(b), 211.188, 211.194, and 212.60). During data acquisition,
for example, pH meters and balances may create a paper printout or static record as the original record. In this case, the paper printout or static record, or a true copy, must be retained (§ 211.180).
The stated regulatory expectation of both the PIC/S and FDA guidance documents for simple analytical instruments is for paper printouts that are attached to an analytical batch record or a laboratory notebook. Whilst there is limited ability to interfere with the printout, without an audit trail and e-records, weighing can be repeated until the “right” result is obtained. Whether a static record is the right approach shall be discussed later in this column.
Static Data are an Integral Part of an Analytical Procedure
Simple instruments such as balances generate data that are critical to the whole of an analytical procedure because the values are inputs to any quantitative chromatographic analysis, for example:
- Determination of purity
- Stability testing
- Impurity determination
- Residual solvent measurement
Here static data become an integral component of a dynamic data process.
However, many laboratories still have inappropriately designed data processes for the small instruments that generate critical data on paper printouts. This is compounded by manually entering the weight into a computerized system or spreadsheet, resulting in transcription error checking by the reviewer.
We can look at this situation further in Figure 1; on the left of the diagram is the generation of a static data record: the balance printout. On the right, weights of samples and standards are manually entered into a CDS that generates dynamic data used to generate the reportable result. Here we can see the conversion of static data into dynamic data. If the analyst entering weights into the CDS mistypes a value that must be corrected later with a corresponding audit trail entry, what was static data on the printout is now dynamic data in the CDS.
This poses a key question:
Why make data static first,only to make it dynamic later?
Is Regulatory Guidance Correct?
The heading poses a fundamental question: Where is the event horizon when considering if data are static or dynamic? Consider the situation on the left side of Figure 1:
- Static Data: Analytical balance and printout in isolation:
Weigh sample + print result = static data - Dynamic Data: The balance and sample weight are integral parts of an overall CDS analytical procedure:
Weigh sample + print result + type weight into CDS + acquire data + generate result = dynamic data
The dynamic equation only takes a high-level view of an analytical procedure, what also must be considered are all the contextual metadata generated throughout the process.
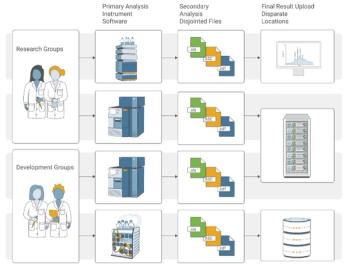
The records generated are a mixture of paper printouts (in yellow) and electronic records (in green) and yet more paper, as shown in Figure 2. Whilst there may be some static data records generated at the start with sample preparation and the balance printout, the overall process generates dynamic data. We now have an analytical procedure that generates hybrid records.
Even if the instrument data system in Figure 2 were to use electronic signatures, only one item of paper would be eliminated (the paper printout on the right of the figure). A hybrid analytical procedure still remains.
Figure 2 shows the complete record set that needs to be reviewed and archived, but the problem is that there are several static paper records plus electronic records.
Are hybrid analytical procedure records an adequate situation to be in?
The one regulatory guidance document that advocates process efficiency is the WHO Good Record and Data Management Practices Guidance from 2016 (3), which states in section 5.6:
A data management programme developed and implemented on the basis of sound QRM principles is expected to leverage existing technologies to their full potential. This, in turn, will streamline data processes in a manner that not only improves data management but also the business process efficiency and effectiveness, thereby reducing costs and facilitating continual improvement.
It is unusual to find a regulatory guidance document recommending business process efficiencies and effectiveness. However, there are two points to be made here:
- Use informatics solutions to streamline processes.
- Eliminate paper and have only a single medium to review and retain.
This is coupled with the statements that “hybrid systems are not encouraged” and “hybrid systems should be replaced at the earliest opportunity” (3). The same approach should be applied to hybrid analytical procedure records because it is not just computerized systems but also processes that require assessments of data vulnerabilities (2,3,5).
However, are static records from simple instruments and the use of hybrid systems acceptable in the third decade of the 21st century? To find the answer we need to travel back to 1978.
Understanding the c in cGMP
Most analytical scientists working in a quality control or analytical development laboratory will refer to GMP regulations; however,
the formal title of 21 CFR 211 is Current Good Manufacturing Practice for Finished Pharmaceutical Products or cGMP regulations.
Is current important? What does current mean?
A question was posed in the introduction of this white paper:
Are the regulators failing to follow their regulations and guidance when it comes to the regulated analytical laboratory? To answer this question, we must go back to 29 September 1978 where the preamble to FDA regulation 21 CFR 211 on Current Good Manufacturing Practice for Finished Pharmaceutical Goods can be found. Preamble comment 17 has the following statements that have been edited for brevity (9):
Several comments objected to the word “current” in the title and text of the regulations…. Several of these comments reflect, the Commissioner believes, a misunderstanding regarding the use of the word “current”….
Although the practices must be “current” in the industry, they need not be widely prevalent. Congress did not require that a majority or any other percentage of manufacturers already be following the proposed mandated practices as long as it was a current good manufacturing practice in the industry e.g. it has been shown to be both feasible and valuable in assuring drug quality.
The takeaway message from this is that laboratories must keep current with technologies and implement new approaches and systems to keep “current”. This is also mirrored in a page on the FDA website (14) that says:
Accordingly, the “C” in CGMP stands for “current,” requiring companies to use technologies and systems that are up to date in order to comply with the regulations. Systems and equipment that may have been “top-of-the‑line” to prevent contamination, mix‑ups, and errors 10 or 20 years ago may be less than adequate by today’s standards.
Is this approach to keeping current unique and only applicable to the FDA?
No, it is mirrored in Article 23, §1 of European Directive 2001/83/EC (10) that requires a marketing authorization holder to:
After a marketing authorization has been granted, the marketing authorization holder shall, in respect of the methods of manufacture and control provided for in Article 8(3)(d) and (h), take account of scientific and technical progress and introduce any changes that may be required to enable the medicinal product to be manufactured and checked by means of generally accepted scientific methods.
Therefore, both FDA and EU regulations expect laboratories to keep up with technologies and be subject to continual improvement as required by ICH 10 on Pharmaceutical Quality Systems (15) and EU GMP Chapter 1 (16).
However, the situation in many laboratories does not match the requirements either to keep current or to take account of scientific and technical progress. In general, a lazy approach is prevalent: if it was good enough last inspection, it will be good enough for the next one. Further arguments may involve citing a lack of money for investment. These cease immediately after receiving regulatory citations when money for remediation flows like water over Niagara Falls.
Apart from the WHO guidance presented above, none of the regulatory guidance documents really consider the requirement for keeping current. This is particularly the situation with PIC/S PI-041 and the FDA quotations cited earlier for simple instruments to print outputs (4,5). Do these guides leave the pharmaceutical industry with a complacent attitude that paper records are OK?
Digitization of the Laboratory
Keeping current with respect to GMP and digitization of a regulated laboratory are two sides of the same coin. Both require automation, computerization, and elimination of paper records that result in complete electronic records in the same informatics system, thus making records and data easy to search and share.
One approach to keeping current with existing technology is shown in Figure 3. The informatics applications used are:
- Laboratory information management system (LIMS), shown in yellow, for managing sample information and collating test results;
- Instrument data system, typically developed by the instrument manufacturer, is interfaced with an analytical balance (interfacing other instrument types is also possible);
- CDS in green;
- Laboratory execution system (LES), in blue, is an option for documenting sample preparation and transferring data to the CDS.
Sample and batch information are transferred to the LIMS and then downloaded to the LES and CDS applications. Sample preparation is automated using the LES and the information, such as dilutions, by the CDS. In Figure 2, the weight from the analytical balance is manually entered into the chromatography data system, which is slow and error-prone. In Figure 3, we have an instrument data system with an electronic workflow that acquires the balance sample weight and transfers it to the CDS. Further technical controls ensure that the right type of balance is used or even a specific instrument identified by its serial number. There is an audit trail to ensure that samples are not weighed into compliance.
The CDS now has all the data required. The sequence is run. Chromatographic data files are interpreted and a reportable result generated. Second person review is accomplished electronically
in the various informatics systems. The scope of review is limited by effective use of validated technical controls within the application, for example:
- No user has deletion privileges, so a reviewer does not need to look for deleted data.
- Data can only be stored in a single location; therefore, no searches need to be conducted for orphan data.
- Audit trails in the applications highlight modified data, allowing a reviewer to review by exception (5).
The informatics applications ensure that all data are complete, consistent, and accurate. This approach also leverages existing technologies as advocated by the WHO data integrity guidance discussed earlier (3),
in addition to meeting the expectation of current in cGMP (9,17).
If required, the values from many results sets can be monitored for trends as required by EU GMP Chapter 6.9 (18) or used to identify errors.
This work would not be possible with static data without extensive manual data entry into a spreadsheet.
The Regulatory Driver for Laboratory Digitization
The FDA are keeping current in cGMP by interpreting the same regulation differently over time. For example, there is no explicit mention of audit trail review in 21 CFR 211 regulations, the bulk of which was issued in 1978 (9). However, since 2005 and the Able Laboratories fraud case (19), the regulatory expectation of the FDA is that audit trail entries must be reviewed. EU GMP has taken a different and better approach by updating Annex 11 for Computerized Systems where there is an explicit requirement for audit trail review of GMP-relevant changes and deletions (20). Audit trail review is now a feature of all regulatory authority data integrity guidance documents (1–5).
Another, less subtle, driver for automation is evidenced by some recent FDA warning letters. In July 2020, FDA issued warning letters to Stason Pharmaceutical (11) and Tender Corporation (12). Although they were cited under different clauses of 21 CFR 211, the extensive remediation required by the FDA was virtually identical for the two companies. A detailed review of these two warning letters and a discussion on understanding the cost of non-compliance is available (21). One specific remediation requirement for both organizations was:
- Technological improvements to increase the integration of data generated through electronic systems from standalone equipment (e.g., balances, pH meters, water content testing) into the LIMS network.
This is a clear message that paper records from standalone systems are not acceptable. The citation also mentions a LIMS network, which can include different informatics applications such as LIMS, LES, ELN, and instrument data systems, as shown in Figure 3.
It is better to acknowledge the problems in your laboratory and have a plan to remediate them at your pace rather than have a tight deadline imposed to appease a regulatory authority. It is much cheaper as well.
However, is interfacing a balance to a LIMS the only way forwards? Connecting the balance to an instrument data system may be a better option as additional metadata can be acquired during the weighing process to ensure the integrity of data. As shown in Figure 3, the instrument data system has the advantage of using the weights from a balance in another instrument workflow. Also, the use of an instrument data system also provides the laboratory with additional resilience if the LIMS is unavailable due to loss of connectivity to the central system or the Cloud.
If an analytical instrument is purchased, how a laboratory uses it can also result in a regulatory citation as BBC Group found out in a warning letter in August 2021 (13):
Your viscometer and UV–vis spectrophotometer had the capability to save data from product/material testing. Despite having this capability, your analysts failed to save the complete, dynamic testing data, and therefore the data was not available for review by the FDA investigator.
If an instrument has the capability to store electronic records and this feature is not used, it can result in a regulatory citation. Equally so, an old instrument operating in a laboratory without the capability to either connect to a printer or store data is a problem. It falls into the FDA argument that it is not top of the line (14), as discussed above.
If a laboratory does not digitize there can be expensive remediation costs, with a timescale determined by the need to convince a regulator that the company is serious about compliance.
Business Driver for Laboratory Digitization
Regardless of the regulations, organizations should consider digitization from a business efficiency perspective alone. Quality control (QC) testing is often at the end of manufacturing and can be seen as slowing the release of a batch of product. If a laboratory was more efficient with analysis and release of the certificate of analysis, what would be the impact on company cash flow if each and every batch were released to the market one day earlier? The way to do this is to work smarter and work electronically through digitization of the laboratory and the removal of paper.
The Covid-19 pandemic has forced laboratories to view a new way of working—some analysts working in the laboratory and others remotely. This can only work if data are electronic and not on paper. You cannot share paper remotely. Imagine doing laboratory work such as second person review remotely in an electronic environment. Review of data and audit trail entries as well as signing results and reports can be performed remotely. Trending can also be performed electronically to meet the requirements of EU GMP Chapter 6.9 (18). You cannot do these tasks with working practices that involve static data and hybrid records.
Conclusion
Some static data are less static than others because the values are manual inputs into a dynamic analytical procedure. Original records from simple instruments must be complete and include all metadata and must be subject to review. However, the review includes transcription error checking, which is a slow and error-prone process.
Regulatory guidance documents on data integrity—with the exception of that from the WHO—do not discuss the need for regulated laboratories to keep current but should do so to be compliant with the regulations that they are supposed to enforce. However, the benefits of managing data in a fully electronic process provides significant business benefits and regulatory compliance, with data stored in one medium. This approach facilitates efficiency and effectiveness including ease of performing data analytics.
This is the way to keep current with cGMP.
Acknowledgement
I would like to thank Gunnar Danielson for helpful comments during the preparation of this column.
References
- MHRA, GMP Data Integrity Definitions and Guidance for Industry 2nd Edition (Medicines and Healthcare Products Regulatory Agency, London, UK, 2015).
- MHRA, GXP Data Integrity Guidance and Definitions (Medicines and Healthcare Products Regulatory Agency, London, UK, 2018).
- WHO, Technical Report Series No.996 Annex 5 Guidance on Good Data and Records Management Practices (World Health Organization, Geneva, Switzerland, 2016).
- US Food and Drug Administration, Guidance for Industry Data Integrity and Compliance With Drug CGMP Questions and Answers (FDA, Silver Spring, Maryland, USA, 2018).
- PIC/S, PI-041 Good Practices for Data Management and Integrity in Regulated GMP / GDP Environments Draft (Pharmaceutical Inspection Convention / Pharmaceutical Inspection Cooperation Scheme, Geneva, Switzerland, 2021).
- GAMP Guide Records and Data Integrity (International Society for Pharmaceutical Engineering, Tampa, Florida, USA, 2017).
- GAMP Good Practice Guide: Data Integrity - Key Concepts (International Society for Pharmaceutical Engineering, Tampa, Florida, USA, 2018).
- GAMP Good Practice Guide: Data Integrity by Design (International Society for Pharmaceutical Engineering, Tampa, Florida, USA, 2020).
- 21 CFR 211, Current Good Manufacturing Practice for Finished Pharmaceuticals, Federal Register, 43(190), 45014–45089 (1978).
- Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use, Official Journal of the European Union 311, 67 (2001).
- FDA Warning Letter Stason Pharmaceuticals, Inc. (2020). Available from: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/stason-pharmaceuticals-inc-604889-07082020.
- FDA Warning Letter Tender Corporation (2020). Available from: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/tender-corporation-599789-07232020.
- FDA Warning Letter BBC Group Limited (2021). Available from: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/bbc-group-limited-614659-08042021.
- Food and Drug Administration, Facts About the Current Good Manufacturing Practices (CGMPs) (2021). Available from: https://www.fda.gov/drugs/pharmaceutical-quality-resources/facts-about-current-good-manufacturing-practices-cgmps.
- ICH, Q10 Pharmaceutical Quality Systems (International Conference on Harmonisation, Geneva, Switzerland, 2008).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 1 Pharmaceutical Quality System (European Commission, Brussels, Belgium, 2013).
- European Union Directive 2001/83/EC on Medicinal Products for Human Use (European Commission, Brussels, Belgium, 2001).
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 6 Quality Control (European Commission, Brussels, Belgium, 2014).
- Able Laboratories Form 483 Observations. 2005. Available from: https://www.fda.gov/media/70711/download.
- EudraLex, Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 11 Computerised Systems (European Commission, Brussels, Belgium, 2011).
- R.D. McDowall, Spectroscopy 35(11), 13–22 (2020).
Articles in this issue
Advertisement
Related Content
Advertisement



Advertisement
Advertisement
Trending on LCGC International
1
LC-MS/MS Links Vitamin D Levels to Sleep Timing
2
HPLC, PTR-ToF-MS Enable AI Grading of White Tea
3
How Should You Respond to FDA 483 Observations?
4
LC-MS/MS Finds PFAS Migration from Stuffed Toys
5