






We provide a high-level overview of Current Good Manufacturing Practice (cGMP) regulations and their impact on the analytical chemist in the pharmaceutical industry.
We provide a high-level overview of Current Good Manufacturing Practice (cGMP) regulations and their impact on the analytical chemist in the pharmaceutical industry.

GLP helps ensure the quality of different studies during drug development. Here, the authors outline practices relevant to analytical and bioanalytical chemists.

“Questions of Quality” is 30 years old! What, if anything, has changed in chromatography laboratories over that time?

The new General Chapter <621> does reflect the great work of the Pharmacopoeias in helping chromatographers to use more modern technology for their regulated methods, speeding up analysis, and reducing waste. The latest revision allows us to adjust column dimensions for gradient separations, which is a significant advance. Although these freedoms are welcomed, one must be very mindful of the requirements or limits of the allowable changes and the verification work that underpins our demonstration of equivalent performance of the adjusted method.

The lack of proper method validation has important implications in blood alcohol concentration (BAC) determinations. We examine this issue, addressing the importance of gas chromatography (GC) for BAC determination, why certain validation procedures are important, and why accreditation bodies need to step up their game.

The advantages of a risk-based approach to instrument replacement are discussed.

The Column
Change, while scary, is a necessity. Opportunities for increased efficiency, higher profitability, and the ability for an organization to maintain a competitive advantage necessitate the need for change. Correctly adopting new liquid chromatography (LC) technology can be very challenging, however, when done correctly, modern LC technology can give a laboratory the ability to simplify method transfer across a diversity of analytical LC platforms at all of its facilities.

LCGC North America
The first three articles in this series discussed where and how a CDS fits into a regulated laboratory, the overall requirements for the architecture of a future system, and additional items to enable effective electronic ways of working. The final part of this series looks at regulatory compliance of a future system as well as a summary of the 15 recommendations made in this series.
The Column
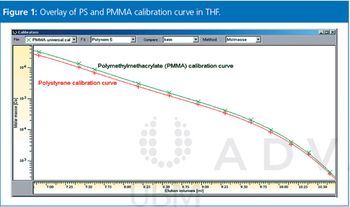
Polystyrenes (PS) are the most commonly used reference standards in gel permeation chromatography/size-exclusion chromatography (GPC/SEC) for nearly all organic GPC/SEC separations including high temperature GPC and pullulan or dextran for aqueous GPC/SEC. The majority of users rely on these standards for reproducible results. However, there is potential for improvements and this instalment of Tips & Tricks will discuss some general points that should be considered when determining calibration standards.

The Column
The fate and modification of nanoparticles in real life matrices can be investigated by the combination of field-flow fractionation (FFF) with inductively coupled plasma mass spectrometry (ICP–MS). This article explains more.

The Column
Calibration using broad standards in GPC/SEC is discussed.

LCGC Asia Pacific eNews
The National Institute of Standards and Technology (NIST), an agency of the U.S. Department of Commerce, has made a polycyclic aromatic hydrocarbon (PAH) structure index database publicly available on-line (http://pah.nist.gov/). A byproduct of hydrocarbon fuel combustion, PAHs can have significant adverse health and environmental impacts. The website contains data on more than 650 PAH compounds, with more to be added in the future.
LCGC Europe eNews
The United States Food and Drug Administration (FDA) and the National Institutes of Health (NIH), as part of an ongoing interagency partnership, have awarded a total of up to $53 million to fund tobacco-related research in fiscal year 2013 to create 14 Tobacco Centers of Regulatory Science (TCORS).
E-Separation Solutions
EMD Millipore, the Life Science division of Merck KGaA of Darmstadt, Germany has been invited to serve on the faculty of the Center for Pharmaceutical Information and Engineering Research (CPIER) at Peking University in Beijing, China.
LCGC Europe eNews
PerkinElmer (Waltham, Massachusetts, USA) has added a Universal Operational Qualification (UOQ) as part of its suite of compliance services for pharmaceutical, biotech and other regulated laboratory environments.

LCGC North America
Here's how these new reference standards were characterized.

LCGC Europe
How will the new revision of EU GMP Annex 11 on Computerized Systems and Chapter 4 on Documentation affect chromatographers?

The Column
Two-dimensional polyacrylamide gel electrophoresis reveals regulatory effects of dietary polyunsaturated fatty acid supply on protein expression in early development.

LCGC Europe
There's more to the data than you might expect.

Special Issues
Common systems for instrument control, data acquisition, and analysis would enhance the productivity of most laboratories greatly.
LCGC Europe
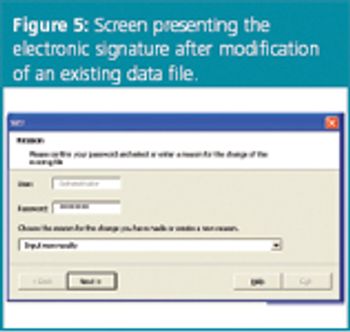
In a good laboratory practice (GLP) environment, data-handling software cannot be used until it is validated. This even applies to the most simple program that performs calculations or stores data. A detailed documentation of the set-up and the performance of the software - called software validation - is required. The development and validation requirements are described in this article and illustrated with a software for robustness testing (SRT), which guides the user step-by-step through the experimental set-up and interpretation of robustness tests. This software was developed in an Excel (Windows XP) environment and is used as part of method validation in laboratories that require compliance with GLP and 21 CFR Part 11. The software was subjected to software validation regulations and is compliant with electronic records and signature rules (21 CFR Part 11) as it creates, delivers and stores electronic data. The validation tests are based on the computerized system validation (CSV) -..

LCGC North America
October 2006. This month's "MS - The Practical Art" will interest those starting a new good laboratory practices (GLP) bioanalytical laboratory, reassessing an existing lab, or revamping a "spirit-of-GLP" laboratory to full GLP status.

Special Issues
FDA is modernizing and streamlining the regulatory processes for product development. This article examines FDA's proposed rule to exempt the production of Phase 1 clinical trial materials from the GMP regulations and questions whether this proposed exemption will truly improve public health and promote faster and more predictable access to new medicines.
LCGC Europe
The authors discuss the issue of meeting the demands of regulatory compliance whilst ensuring good scientific practice. A number of requirements from 21 CFR Part 11 are cited to demonstrate the importance of applying the principles of risk analysis.