
|Articles|September 1, 2019
- LCGC Europe-09-01-2019
- Volume 32
- Issue 9
Clinical Metabolomics: Expanding the Metabolome Coverage Using Advanced Analytical Techniques
This review article discusses the novel separation and detection strategies that are considered promising in clinical metabolomics to enhance the metabolome coverage. It includes hydrophilic interaction chromatography (HILIC), supercritical fluid chromatography (SFC), multidimensional LC approaches, as well as ion-mobility mass spectrometry (IM-MS) and data-independent acquisition (DIA) analysis methods.
Advertisement
Metabolomics, the comprehensive analysis of all metabolites and intermediate products of reactions present within a biological system, is a promising field to enable precision medicine. Clinical metabolomics faces two main challenges at the bioanalytical level. The first is the need for high resolution to obtain maximum metabolome coverage. This is exemplified by the latest version of the Human Metabolome Database (HMDB), which reports more than 110,000 metabolites and endogenous compounds. The second is the high-throughput needed to enable the analysis of a large number of samples typically encountered in large-scale cohort studies. Reversed-phase liquid chromatography (LC) at regular or ultrahigh pressures combined with high-resolution mass spectrometry (HRMS) has long been considered the “gold standard” in metabolomics. However, these conventional reversed-phase LC–MS approaches are no longer sufficient to analyze the vast variety of polar compounds, as well as discriminate closely related compounds such as isomers or enantiomers. This review article discusses the novel separation and detection strategies that are considered promising in clinical metabolomics to enhance the metabolome coverage. It includes hydrophilic interaction chromatography (HILIC), supercritical fluid chromatography (SFC), multidimensional LC approaches, as well as ion-mobility mass spectrometry (IM-MS) and data-independent acquisition (DIA) analysis methods.
Metabolomics, first formally introduced in the early 2000s and described as the comprehensive analysis of all metabolites present within a biological system, has attracted a growing interest over the last decade in clinical research. Together with other “omics” approaches, such as genomics and proteomics, metabolomics plays a key role in the implementation of personalized medicine. Two approaches are typically considered in metabolomics. In targeted metabolomics, known metabolites from given biochemical pathway(s) are measured in a quantitative manner. Untargeted approaches, on the other hand, focus on the global and unbiased analysis of the highest number of compounds included in the metabolome leading to qualitative and semiquantitative information (relative differences between populations). Both approaches have been increasingly used over the last couple of years in personalized medicine and drug discovery, in the aim of finding new metabolite biomarker candidates for earlier and more accurate diagnosis, improve the prognosis and staging of diseases, and increase the global understanding of pathophysiological processes via the discovery of novel biomolecular pathways (1,2).
In 2017, the fourth version of the Human Metabolome Database (HMDB 4.0) covered more than 110,000 fully annotated metabolites. This is a threefold increase compared with the previous release of HMDB 3.0 in 2013 (3). The human metabolome is very complex and comprises a large diversity of compounds, including amino acids, organic acids, nucleosides, lipids, small peptides, carbohydrates, biogenic amines, hormones, vitamins, and minerals. Moreover, xenobiotics such as drugs, cosmetics, contaminants, pollutants, and their respective phase-I and phase-II metabolites are also part of this metabolome. The (ideally) comprehensive analysis of the metabolome is therefore linked to several analytical challenges due to (i) the large differences in physicochemical properties (polarity, solubility, pKa values, molecular mass), (ii) the broad dynamic range needed to analyze both trace compounds and highly abundant metabolites (up to nine orders of magnitude difference), and (iii) the presence of multiple isomers with structural similarities but significant differences in their biological activities (lipid-based signalling molecules) (4).
Overall, this complexity highlights the need for stateâofâtheâart analytical approaches capable of tackling such challenges and enabling a qualitative and quantitative assessment of the metabolome. This should be done with the highest possible metabolic coverage via high resolving power and selectivity. In this context, metabolomics has strongly benefitted from the latest developments in the fields of both chromatography and mass spectrometry (MS) over the last two decades. The use of reversedâphase liquid chromatography (LC) columns equipped with sub-2-µm fully porous particles (ultrahighâpressure liquid chromatography, UHPLC) or sub-3-µm superficially porous particles (core–shell technology) are now considered wellâestablished methods in metabolomics owing to the dramatic improvements in resolution and throughput obtained with such phases compared with conventional high performance liquid chromatography (HPLC) (5–7). On the other hand, recent liquid-based chromatographic and MS innovations, notably within hydrophilic interaction chromatography (HILIC), supercritical fluid chromatography (SFC), multidimensional LC, ion-mobility mass spectrometry (IM-MS), and data-independent acquisition (DIA) approaches, are not widely used in metabolomics, despite the significant improvement in metabolite coverage expected with such techniques. Therefore, in this review article, the latest developments in the aboveâmentioned fields of chromatography and MS are discussed with a focus on their ability to increase the metabolome coverage.
Improvement of Metabolic Coverage: Chromatographic Innovations
Hydrophilic Interaction Chromatography: Reversed-phase LC-based methods have long prevailed in metabolomics because of the large variety of column chemistries available, the ease of use, and retention time reproducibility. However, a large number of polar or ionizable metabolites, such as amino acids, small organic acids, nucleosides, phosphate derivatives, or saccharides, are not well-retained using reversed-phase LC. Still, many of these polar metabolites play an essential role in multiple (patho)physiological processes, showing the need for alternative approaches. HILIC, a technique first proposed by Alpert in 1990 (8), is well-suited for the analysis of polar compounds. Retention is based on a multimodal separation mechanism between a polar stationary phase and a relatively hydrophobic mobile phase composed of an aqueous–organic mixture with a high organic proportion. With a concentration of 5–40% of water in the eluent, a water-enriched layer is formed at the surface of the stationary phase, facilitating analyte partitioning between this stagnant phase and the bulk mobile phase. The exact mechanisms involved in retention and separation are not fully understood but mostly rely on hydrophilic partitioning, dipole-dipole interaction, hydrogen bonds, and electrostatic interactions (depending on the stationary phase chemistry) (5,9).
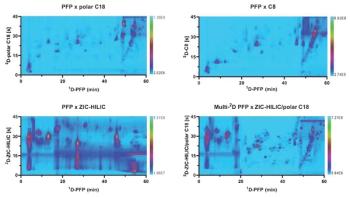
A large diversity of phase chemistries based on silica or polymer material modified with polar functional groups, for example, aminopropyl and amine, amide, diol, triazole, sulfobetaine, phosphorylcholine, hydroxyethyl, and sulfoethyl are nowadays commercially available for HILIC analysis. Whereas for reversed-phase LC analyte retention can be easily predicted, helping to facilitate method development, this remains difficult in HILIC. The chromatographic selectivity is also strongly dependent on the stationary phase chemistry and composition of the mobile phase, as illustrated in Figure 1. A careful and extensive screening of different conditions during method development using a large set of representative metabolites is therefore recommended to obtain an adequate metabolite coverage. The help of modern computer-assisted method development strategies, such as the predictive elution window shifting and stretching (PEWS2) approach (10), could be useful here to speed up method development. Numerous studies comparing the different stationary phases for metabolomics applications showed that diol, amide, and zwitterionic phases usually give the best results in terms of metabolite coverage, therefore representing a good starting point in the method development process (11,12). Small organic acids, sugar phosphates, and nucleosides are difficult to analyze with reversed-phase LC. Using HILIC mode these compounds are better retained, especially with polymeric zwitterionic phases, which allows analysis at high pH (pH 9–10) thanks to the polymeric nature of the stationary phase (13). Adding phosphate at micromolar concentrations to the mobile phase has also shown to further improve the peak shape and sensitivity when analyzing such metabolites with a zwitterionic phase (14). Next to the stationary phase chemistry, the composition of the mobile phase strongly influences the selectivity and quality of the separation. Acetonitrile is the optimal organic solvent because it is water-soluble and aprotic. Protic solvents such as methanol, isopropanol, and ethanol are not recommended due to competition with water for the solvation of the stationary phase, which may lead to lower analyte retention (15,16). In HILIC, the high proportion of acetonitrile in the mobile phase decreases its viscosity compared with the mobile phase mixtures used in reversed-phase LC, which offers additional advantages such as the possibility to use longer columns (leading to higher efficiencies), a higher electrospray ionization (ESI) sensitivity, and higher volatility (15). The buffer composition (that is, salts concentration and pH) has a strong impact on both selectivity (Figure 1) and retention time reproducibility. The buffer concentration (commonly ≤50 mM to avoid salt precipitation in acetonitrile) influences the thickness of the water layer and thus the hydrophilic interaction, and plays an essential role in electrostatic interactions. Ammonium formate and acetate buffers are commonly used because of their MS compatibility. They also give better peak shapes than the corresponding acid solutions (16). Trifluoroacetic acid is not recommended in HILIC–MS because it leads to strong ion suppression in the range of compounds studied. Finally, an adequate and repeatable buffer pH is crucial in HILIC to ensure reproducible analyses. Changes in buffer pH will lead to a higher retention variability, showing the importance of repeatable procedures when preparing the buffer solutions.
Despite all the above mentioned advantages and the improved metabolite coverage that can potentially be obtained using the technique, HILIC remains sparsely used in metabolomics, mostly confined to untargeted studies (16,17). The complex mechanism of HILIC separation, the longer equilibration times, the attention required to ensure reproducibly prepared mobile-phase buffers, and the challenges in finding an adequate sample injection solvent might explain why this technique has not been widely adopted yet. However, there are now numerous excellent reviews available discussing these challenges, offering solutions and providing guidelines for state-ofâthe-art HILIC analysis (1,9,15,16,18). This will hopefully foster the use of HILIC in routine metabolomics.
Supercritical Fluid Chromatography: Although not new-the use of fluids in their supercritical state was first reported in the 1960s-SFC has shown a spectacular comeback in the last decade. This is due to the introduction of a new generation of instruments capable of performing robust, reproducible, reliable, and quantitative analysis. Similar to what has been observed in conventional LC, these new instruments have also fostered the development of columns packed with subâ2âµm fully porous (ultrahighâperformance SFC, UHPSFC) and sub-3-µm superficially porous particles specially designed for SFC analysis. Moreover, the new source designs recently developed for interfacing SFC with MS have also strongly contributed to developing the use of SFC in bioanalysis, including metabolomics (19,20). The metamorphosis of the technique has transformed UHPSFC–MS into a very competitive separation approach, complementary to UHPLC–MS, as underlined in the first ever inter-laboratory study. Between the 19 participating laboratories, similar or even better repeatability and reproducibility using SFC was shown for the determination of impurities in pharmaceutical formulations compared with conventional LC methods (21).
Supercritical fluids have unique properties that take advantage of both gas and liquids, with viscosity and diffusivity very close to those of a gas, while their density and solvating power is close to those of a liquid. Overall, these inherent characteristics enable high separation efficiency at high mobile phase velocity with a low back pressure generated, and good solvation and fast transportation of the analytes (22). Carbon dioxide has been considered the solvent of choice as a result of the low critical temperature and critical pressure (31 °C and 74 bar, respectively) as well as its low toxicity, low flammability, and environmentally friendly properties. However, the low polarity of pure CO2 limits its application to the analysis of rather nonpolar or hydrophobic compounds such as lipids. The addition of a miscible co-solvent (referred to as modifier, typically methanol) to the mobile phase is an adequate strategy to enable the retention of polar compounds. The use of an organic coâsolvent influences the solvating power of the mobile phase, its hydrogen-bonding donor and acceptor properties, its density, the interaction between analytes and mobile phase, as well as the adsorption of analytes on the stationary phase (20,22). Yet, adding a modifier to this supercritical fluid increases the critical temperature and pressure of the fluid. In current applications, the pressure is commonly maintained over its critical point while the temperature is below its critical value. In this case, the fluid is in a subcritical state, showing a chromatographic behaviour close to LC.
The addition of acids (formic acid, citric acid), bases (trimethylamine, isopropylamine), or salts (ammonium acetate, ammonium fluoride) at low concentrations in the modifier also increases the range of compounds that can be analyzed using SFC, especially ionizable compounds such as polyacids, aliphatic amines, and other polar metabolites (20,22). These additives also increase the separation efficiency and peak shape by acting as ionâpairing agents and by covering active sites on the stationary phase, leading to less tailing and better elution of polar compounds. The latest trend in SFC is the use of water as an additive in a CO2–methanol mobile phase to improve peak shape (at a proportion of 1–5%, miscible in the mobile phase) or to enable the elution of very polar compounds (up to 30%, forming a ternary mixture) (20,22).
A number of SFC-specific stationary phases have been developed and commercialized in the past years, including 2-ethylpyridine, 4-ethylpyridine, pyridine amide, amino phenyl, 2-picolylamine, diethylamine, diol, and 1-aminoanthrocene. In addition to SFC-specific columns, reversed-phase LC- and HILICâtype stationary phases (ethyleneâbridged silica, C18, fluorophenyl, amide) can also be used in SFC.
Most of the SFC-specific columns are also available in sub-2-µm format. However, the extracolumn band broadening of the state-of-the-art UHPSFC instruments currently on the market are still higher than the corresponding values obtained on UHPLC systems (namely, 85 µL2 vs. 2 to 20 µL2), hindering the use of typical UHPLC column dimensions (50 mm × 2.1 mm, 1.7-µm) with these systems. On the other hand, 4.6-mm internal diameter (i.d.) columns require flow rates above the system limits (22). Therefore, most of the current state-of-the-art SFC applications are performed using 100 mm × 3.0 mm columns packed with sub-2-µm fully porous and sub-3-µm superficially porous particles, which represents an adequate compromise and can lead to excellent kinetic performance with a low pressure drop, as illustrated in Figure 2 (23).
Modern UHPSFC–MS analysis has recently started to gain more attention from the metabolomics community, not only in the fields of lipidomics but also as a complementary technique to UHPLC–MS to increase the metabolome coverage. Multiple metabolite classes, including amino acids, bile acids, cannabinoids, fatty acids, saccharides, steroids, and tocopherols, have been successfully analyzed using UHPSFC–MS (24). A good example of the potential of UHPSFC was described by Holcapek and co-workers, who demonstrated the comprehensive and quantitative analysis of different lipid classes (25). Figure 3 shows the chromatogram obtained for the analysis of lipid internal standards with UHPLC–HILIC–MS (Figure 3[a]) and with UHPSFC (Figure 3[b]), both coupled to a quadrupole–travelling-wave ion mobility–time-of-flight (TOF) mass analyzer. Both HILIC and UHPSFC enable the separation of lipid classes without the typical overlap that is seen when using conventional reversedâphase LC approaches. Nonpolar lipids (cholesterol esters and triglycerides) as well as species with one hydroxyl group (ceramides, diglycerides, monoglycerides, and cholesterol) show poor retention in HILIC and elute in the void volume (Figure 3[a]). On the other hand, all lipids are retained using UHPSFC (Figure 3[b]). Whereas the positional isomers 1,2-DG/1,3-DG and 1,2-MG/1,3-MG are well resolved in UHPSFC (Figure 3[b]), the positional isomers of the more polar lysolipids 1-LPG/2-LPG, 1-LPE/2-LPE, and 1-LPC/2-LPC are well resolved using HILIC (Figure 3[a]) (25). This example illustrates the complementary nature of the different chromatographic modes, where no single analytical technique currently enables a comprehensive coverage of the metabolome.
Despite the excellent performance that can be reached with modern UHPSFC–MS, it remains little used in metabolomics-and only by a limited number of research groups. This reluctance might be explained by the large diversity of stationary phases currently available, together with the flexibility offered in the composition of the mobile phase (modifier, additives, gradient composition). Indeed, the method development step a priori may be seen as very cumbersome and time-consuming. However, it can be facilitated by using column classification maps to help select adequate column chemistries (26) and by using method optimization work (27).
Overall, there is no doubt that the multiple advantages of modern SFC will foster its use in metabolomics in the coming years as a complementary chromatographic approach to expand the metabolome coverage. The advantages of SFC in metabolomics include (i) its application range versatility, with a large range of metabolites with very diverse physicochemical properties that can be analyzed within a single run (as shown in reference 27), (ii) the sample compatibility with the mobile phase used in SFC, (iii) the excellent sensitivity of UHPSFC–MS, comparable or superior to that of UHPLC–MS, and (iv) the flexibility offered with the state-of-the-art instruments, which allows for both UHPLC and UHPSFC analysis within one single system and an unlimited combination of solvent and stationary phases. SFC technology has faced the same reluctance as HILIC a decade earlier, but both techniques are promised to rise further in the field of metabolomics.
Multidimensional Chromatographic Separations: A straightforward approach to increase the metabolome coverage of very complex samples or closely related metabolites is to add another separation dimension to provide additional selectivity. Similar to what has been observed in the field of SFC, onâline two-dimensional liquid chromatography (2D-LC) is far from being a new concept but has seen a significant breakthrough in the last couple of years thanks to significant advances in theory and instrumentation. In on-line 2D-LC, two individual LC separations are combined, typically using a four-port duo valve or 10âport valve with two sampling loops, or connected to special valves with multiple sample parking loops.
Capturing all peaks-or a large number of fractions-from the first dimension into the second dimension is referred to as comprehensive 2D-LC (also called LC×LC), while multiple heart-cutting 2D-LC (also called LC-LC) is used when one or few distinct fractions are collected from the first dimension and are subjected to a high-resolution analysis in the second dimension. Selective comprehensive 2D-LC (sLC×LC) is an intermediate approach where a series of fractions across one or more regions in the first dimension chromatogram are transferred to the second dimension (28,29). The comprehensive LC×LC approach appears particularly interesting in untargeted metabolomics, where hundreds of features can be profiled during one single analysis.
A large diversity of chromatographic modes can be combined in 2D-LC, including reversed-phase LC, HILIC, normal-phase LC, ion-exchange chromatography (IEX), ion-pairing chromatography, and porous graphitized carbon (PGC) columns. Different stationary phase chemistries and mobile-phase compositions can be employed, aiming for the highest orthogonality of separation between the two dimensions. The selection of the two separation dimensions depends on the analytes, the compatibility and miscibility of the mobile-phase solvents, the compatibility with the detector, and the selection of a faster technique (that is, based on UHPLC conditions) for the second dimension (4,28).
With the recent advent of state-of-the-art instruments for 2D-LC analysis, the number of experimental parameters that can be optimized during method development has dramatically increased. Indeed, setting up a complete 2D-LC method requires optimization of multiple parameters, including column dimension, stationary phase, particle sizes, mobile-phase composition, gradient conditions, sample loop volume, injection volumes, flow rates, and modulation times. This can lead to a rather cumbersome and lengthy method development. Moreover, state-of-the-art 2D-LC analyses usually require a dedicated instrument (even though one-dimensional [1D]-LC systems can be upgraded to 2D-LC with only minor investment). Finally, hyphenating 2D-LC to MS adds another challenge, since the insertion of an additional LC dimension may induce a significant dilution of the effluent injected to the MS system (28,30).
Overall, this might explain the reluctance in using this technique in metabolomics, despite the remarkable promises 2D-LC holds in significantly expanding the metabolome coverage. This reluctance is similar to the one observed for HILIC and SFC in metabolomics, where inexperienced users are struggling to get reproducible data. Moreover, they might lack sufficient theoretical and practical knowledge to get the best out of those techniques. However, 2D-LC is currently a very dynamic field and a number of excellent guidelines and tutorials have recently been published by experts in the field, guiding the inexperienced user through this method development (28–31). The recent developments in instrumentation, including the use of active-modulation techniques to alleviate the MS detector sensitivity problems and minimize effects from poorly compatible mobile phases, software tools to support method development, as well as continuous improvements in the algorithm available for processing 2D chromatograms, will certainly foster its use in clinical metabolomics. Most of the applications reported so far have been mostly based on heart-cutting approaches and proof-of-concept studies rather than clinical applications. However, the results presented highlighted the potential of 2D-LC in metabolomics, showing for instance, a twofold increased coverage of intracellular energy metabolites using a combination of reversed-phase LC with PGC (32) and the acquisition of both metabolomic and lipidomic information in a single analysis using heart-cutting 2D-LC (33).
Improvement of Metabolic Coverage: MS Developments
Ion-Mobility Mass Spectrometry: Among all recent developments discussed here, IM-MS is probably the one that has already been largely accepted by the metabolomics community even though it remains a relatively young technique. IM-MS adds an orthogonal separation dimension between chromatographic separation and MS detection without impacting the analysis time. IM-MS separation occurs in a timescale of milliseconds, which makes this technique fully compatible with both fast LC and high-throughput MS approaches (especially TOF mass analyzers, which offer fast duty cycles) (34).
IM-MS is a gas-phase technique separating ions driven through an ion mobility cell under an electric field in the presence of an inert buffer gas. Ions are separated according to their mobility or drift time, which is intrinsically linked to their size, shape, and charge. Assuming that the experimental parameters (for example, drift-tube length, gas pressure, temperature, electric field) are constant, the ion drift time is proportional to the rotationally averaged collision crossâsection (CCS) value, which represents the effective area involved in the interaction between an ion and the gas present in the ion mobility cell. The CSS value is not only highly reproducible but also unique for each analyte, and reflects its chemical structure and three-dimensional configuration. This shows the power of IM-MS in metabolomics, especially in untargeted metabolomics, where CCS values can be used in addition to conventional parameters typically reported in libraries (retention time, mass-to-charge ratio, fragmentation pattern) for metabolite characterization and to increase the confidence in metabolite identification (4,35).
Different IM-MS technologies are currently commercially available, namely, (i) drift-tube ion-mobility spectrometry (DTIMS), (ii) travelling-wave ion-mobility spectrometry (TWIMS), (iii) field-asymmetric ion-mobility (FAIMS), also called differential-mobility spectrometry (DMS), (iv) differential mobility analyzer (DMA), and (v) confinementâandâselectiveârelease ion mobility, also called trapped ion mobility spectrometry (TIMS). They differ amongst each other in terms of applied electric field and state of the buffer gas. DTIMS and TWIMS belong to the timeâdispersive methods, where all ions drift along the same pathway and have a different drift time. FAIMS and DMA are spaceâdispersive methods that separate ions following different drift paths, based on their mobility difference. In TIMS, the ions are first trapped in a pressurized region before being selectively released based on their mobility differences. By using DTIMS instruments, CCS values can be directly derived from the drift time while other approaches require the use of calibrants with known CCS values to calculate the CCS value from the drift time of an unknown (35,36).
IM-MS is able to improve the metabolome coverage by enhancing the selectivity and resolution between metabolites, but one of its major impactful applications probably lies in the field of lipidomics. Indeed, lipid analysis remains exceptionally challenging because of their structural diversity and the multiple lipid isomers that can be present in a biological sample. Contrary to conventional MS/MS approaches, IM-MS enables the discrimination between lipid isomers that differ only in the position of the acyl chain or the double bond, or with a different double bond geometry. An example is shown in Figure 4 with the two lipids 5S,12S-diHETE and LTB4 , both of which arise from different pathways and have different biological activities. 5S,12SâdiHETE and LTB4 are diastereomers and geometrical isomers. Therefore, they show identical mass spectra and similar retention behaviour using conventional LC–MS/MS analysis. However, adding IM-MS (in this example DMS) enables a baseline separation of these two compounds using two different compensation voltages (37).
Beside lipid analysis, IM-MS has also demonstrated its usefulness for the analysis of polar metabolites in various body fluids. Most of these applications were untargeted, as discussed in references 34 and 36.
Despite its promising contribution to improve the metabolome coverage and metabolite annotation using the CCS value, IM-MS still faces important challenges linked to data interpretation. Indeed, in an LC–IM-MS workflow, the potential in-source fragments, dimers, and adducts will also be separated in the ion mobility cell. A correct regrouping and assignment of these signal features adds another layer of complexity, which is currently not completely tackled by the software available, especially in untargeted metabolomics workflows (35).
Data-Independent Acquisition: Another MS-based strategy used to improve the metabolic coverage is dataâindependent acquisition (DIA), which allows for the detection and identification of lower abundant metabolites otherwise not recorded with conventional data-dependent acquisition methods. DIA approaches are not new but they have gained more attention since the advent of SWATH-MS approaches. In DIA, precursors selection windows are defined in the first quadrupole (MS1) of a tandem mass spectrometer; all ions are then fragmented in the collision cell and collected into a composite spectrum in the third quadrupole (MS2). Several DIA techniques have been reported so far, including MSEverything (MSE), all ion fragmentation (AIF), MSX, and SWATH (38). In MSE and AIF, all coeluted precursor ions in the whole selected mass range are fragmented to acquire MS2 spectra. MSE alternatively acquires the full MS1 scan with low collision energy (full MS spectrum) and MS2 scan from all precursor ions with high collision energy (MS/MS spectrum). In AIF, all precursor ions are transmitted into a higher energy collisional dissociation cell for fragmentation. Both AIF and MSE acquisitions generate highly complex multiplexed MS2 spectra. SWATH, which stands for Sequential Window Acquisition of all THeoretical fragment ion spectra and was first described in 2012, has been developed to reduce this data complexity by using a narrow isolation window (39). In SWATH-based DIA techniques, implemented on quadrupole-time-of-flight (QTOF) or less frequently Q-Orbital trap instruments, all precursors ions are sequentially fragmented in a serial of quadrupole isolation windows (Q1 windows). The complete “snapshots” of all metabolite ions and their product ions in MS2 are recorded through the whole chromatogram. The full mass range can be covered in one cycle depending on the selected MS1 scan range and the width of the isolation window. The SWATH windows can be both fixed (typically 25 Da) or variable (that is, the window width is not uniform), and are selected depending on the selectivity required and the cycle time (as short as possible if combined with UHPLC). The complexity of the multiplexed MS2 spectra is therefore decreased by reducing the number of simultaneously fragmented precursor ions, which also improves the overall quantitative performance (37,38).
SWATH-MS is now widely used in proteomics and has emerged as a powerful technique in other clinical applications because of its reproducibility, speed, compound coverage, and quantitation accuracy. The great performance observed in proteomics fields has also attracted the attention of the metabolomics community looking to expand the information gathered on the metabolome within a single run. A number of metabolomics and lipidomics applications of LC–SWATH-MS have already been reported in the literature. For example, UHPLC–SWATH-MS was used to investigate the changes in the urinary metabolome of rat models upon administration of vinpocetine. Information on both drug metabolism and endogenous metabolite expression changes were gathered, with the simultaneous detection of 28 drug metabolites as well as altered endogenous compounds (40). Using a combination of SWATH-MS and selected reaction monitoring (SRM), Zha et al. developed a two-step workflow to discover potential biomarkers for colorectal cancer. In this method, SWATH-MS was first used to acquire the MS2 spectra for all metabolites in one pooled biological sample. In the second step, a large set of SRM transitions was acquired, targeting both known and unknown compounds (around 1000–2000 metabolites). This approach increased the coverage in targeted metabolomics analysis, where more than 1300 metabolite were profiled in one run in colorectal cancer tissues (41).
Further developments of SWATH technology are still required, particularly in the data analysis pipeline. Indeed, in a DIA-based dataset, the direct connections between precursor and product ions are missing, rendering the metabolite identification very challenging. Chromatographic ion profiles can be used to reconstruct these connections, but coelution and co-fragmentation of precursor ions makes it complicated. Several software tools have been recently developed to overcome the challenges related to DIA-based data analysis (39). The open-source software MS-DIAL, for example, uses a mathematical deconvolution of fragment ions to extract the original spectra and reconstruct the link between precursor and product ion, allowing for compound identification, annotation, and quantitation. It also implements additional functions typically used in untargeted data processing, namely, peak alignment, filtering, and missing value interpolation (42).
Overall, SWATH-MS represents a great tool to expand the metabolome coverage and obtain both qualitative and quantitative information within a single run. The complexity of the generated data remains a challenge since the reconstructed spectral quality impacts both the confidence in metabolite annotation and quantitation accuracy. The addition of IM-MS in LC–SWATH-MS workflows might help to decrease the spectral complexity by adding an additional separation of the precursor ion to help facilitate the spectral deconvolution, as well as providing CCS values to help in metabolite identification, but also increase the need for adequate data processing software tools.
Conclusions
The last decade has seen a tremendous amount of technological developments in liquid-phase chromatography and MS techniques, developments initially for other applications but showing a considerable potential in metabolomics. Modern clinical metabolomics applications rely on two essential aspects, namely, high-throughput analysis and comprehensive metabolome coverage. The latter is crucial in the quest for the Holy Grail, that is, the discovery of new biomarkers that could ultimately lead to a better understanding of (patho)physiological conditions, an earlier disease diagnosis, a better prognosis evaluation, and an individualized prediction of treatment response. The more comprehensive the metabolome coverage is, the higher the chances are of finding specific metabolites or metabolite fingerprints.
The chromatographic and mass spectrometric innovations presented here have also largely demonstrated their relevance in expanding the metabolome coverage. Most of those techniques, however, are still in their infancy in the field of clinical metabolomics and are rarely used for large-scale studies, where reversed-phase LC–MS and gas chromatography (GC)–MS remain the gold standard techniques. Very few studies have reported the use of HILIC–MS for the analysis of hundreds of samples, while the robustness of SFC and 2D-LC needs to be further investigated, as well as the potential of these two novel techniques in largeâscale metabolomics applications. One of the main obvious reasons is the lack of practical background knowledge of non-experienced users, who struggle to get repeatable and reproducible results. In this context, leading experts in these fields and professors play a crucial role and are strongly encouraged to share their knowledge with the younger generation of scientists. Moreover, further technological improvements are needed to ensure the batchâto-batch reproducibility of SFC and HILIC chromatographic columns, which currently remains a clear bottleneck in metabolomics. As an example, acceptable repeatabilities can be obtained in HILIC with >1000 injections (depending on the stationary phase chemistry) of pretreated biological samples using standard procedures (including adequate column reâequilibration time). However, it is much more difficult to reach such repeatability when using HILIC columns from different batches, rendering the use of HILIC in large-scale studies much more challenging than reversed-phase LC.
Increasing the metabolome coverage does not stop at discriminating metabolites with close physicochemical properties. An important aspect often overlooked in clinical metabolomics is the distinction of optical isomers (stereoisomers such as enantiomers). An excellent example is 2-hydroxyglutarate (2-HG), the first oncometabolite (cancerâcausing metabolite) ever reported. Both D- and L- stereoisomers of hydroxyglutaric acid are normal endogenous metabolites found in human body fluids. D-2âHG-not L-2-HG-is produced in the presence of gainâof-function mutations of isocitrate dehydrogenase, causing a cascading effect in the cell that leads to genetic perturbations and malignant transformation. Typical routine analytical techniques only measure 2-HG, which strictly speaking corresponds to the sum of both D- and L-forms. Since the endogenous serum levels of L-2-HG have shown to be equal or even exceed the levels of D-2-HG in healthy individuals, it is essential to use stateâof-the-art analytical techniques to discriminate between the two stereoisomers (43). Some of the advanced techniques discussed here, mostly SFC, are applicable to chiral analysis and are therefore expected to play a crucial role in next-generation metabolomics.
Overall, despite the technological improvements within each of the discussed techniques, none of the stateâof-theâart analytical techniques is currently capable of exhaustively assessing the metabolome. SFC will certainly become a gold standard chromatographic technique complementary to reversed-phase LC and HILIC because of the versatility and flexibility offered (convergence chromatography) and the experimental conditions, where a large diversity of metabolites can be analyzed without strong variations of the operating parameters. Moreover, the higher throughput obtained with UHPSFC is also a clear advantage in clinical metabolomics, notably with the next generation of instruments, allowing for higher back pressure to be generated, which is still a limitation in the instruments currently on the market.
The future of metabolomics probably relies on the combination of different separation dimensions in an on-line format, as demonstrated with 2D-LC approaches. The first 2D-LC-SFC application has been reported in the literature for simultaneous achiral-chiral analysis of pharmaceutical compounds (44), a multidimensional approach that might be further investigated for metabolomics-based applications. The combination of multidimensional LC with IM-MS has a promising future in metabolomics, showing the remarkable advantage of improving the metabolome coverage while keeping similar throughput. Alternative approaches based on miniaturization of conventional LC techniques and the use of micro-pillar array columns instead of columns packed with porous particles will also probably help to further expand the metabolome coverage, as already shown in lipidomics where structural lipid isomers were chromatographically baseline resolved using micro-pillar array columns (45).
One should also keep in mind the challenges associated with a substantial improvement of the metabolome coverage. First, the development of cutting-edge analytical instruments should not forget the importance of sample preparation, which should be as simple and generic as possible while providing sufficient clean-up to lower the occurrence of matrix effects. Moreover, an increased number of metabolites in quantitative targeted metabolomics means an increased number of internal standards, which raises the overall costs. Finally, enhancing the number of metabolite features measured in a studied population requests a much higher number of samples and subjects included in the study design to keep a sufficient statistical power, which in turn substantially increases the costs and the number of samples analyzed. A compromise between all aspects is definitely needed to achieve successful results in the field of clinical metabolomics.
References
- I. Kohler, A. Verhoeven, R.J. Derks, and M. Giera, Bioanalysis8, 1509–1532 (2016).
- C.H. Johnson, J. Ivanisevic, and G. Siuzdak, Nat. Rev. Mol. Cell. Biol.17, 451–459 (2016).
- D.S. Wishart et al., Nucleic Acids Res.46, D608–D617 (2017).
- K. Ortmayr, T.J. Causon, S. Hann, and G. Koellensperger, TrAC82, 358–366 (2016).
- I. Kohler and M. Giera, J. Sep. Sci. 40, 93–108 (2016).
- H.G. Gika, G.A. Theodoridis, R.S. Plumb, and I.D. Wilson, J. Pharm. Biomed. Anal. 87, 12–25 (2014).
- P. Rainville, G. Theodoridis, R.S. Plumb, and I.D. Wilson, TrAC61, 181–191 (2014).
- A.J. Alpert, J. Chromatogr.499, 177–196 (1990).
- L. Novakova, L. Havlikova, and H. Vlckova, TrAC63, 55–64 (2014).
- E. Tyteca, A. Periat, S. Rudaz, G. Desmet, and D. Guillarme, J. Chromatogr. A1337, 116–127 (2014).
- S. Wernisch and S. Pennathur, Anal. Bioanal. Chem.408, 6079–6091 (2016).
- D.P. Kloos, H. Lingeman, W.M. Niessen, A.M. Deelder, M. Giera, and O.A. Mayboroda, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 927, 90–96 (2013).
- A. Teleki and R. Takors, Methods Mol. Biol. 1859, 185–207 (2019).
- J.L. Spalding, F.J. Naser, N.G. Mahieu, S.L. Johnson, and G.J. Patti, J. Proteome Res.17, 3537–3546 (2018).
- D. McCalley, LCGC Europe32(3), 114–125 (2019).
- I. Kohler, R. Derks, and M. Giera, LCGC Europe30(2), 60–75 (2016).
- D.Q. Tang, L. Zou, X.X. Yin, and C.N. Ong, Mass Spectrom. Rev. 35, 574–600 (2016).
- J. Ruta, S. Rudaz, D.V. McCalley, J.L. Veuthey, and D. Guillarme, J. Chromatogr. A1217, 8230–8240 (2010).
- D. Guillarme, V. Desfontaine, S. Heinisch, and J.L. Veuthey, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.1083, 160–170 (2018).
- A. Tarafder, TrAC81, 3–10 (2016).
- A. Dispas et al., J. Pharm. Biomed. Anal. 161, 414–424 (2018).
- V. Desfontaine, D. Guillarme, E. Francotte, and L. Novakova, J. Pharm. Biomed. Anal. 113, 56–71 (2015).
- A. Grand-Guillaume Perrenoud, J.L. Veuthey, and D. Guillarme, J. Chromatogr. A 1266, 158–167 (2012).
- V. Shulaev and G. Isaac, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.1092, 499–505 (2018).
- M. Lisa, E. Cifkova, M. Khalikova, M. Ovcacikova, and M. Holcapek, J. Chromatogr. A1525, 96–108 (2017).
- C. West, E. Lemasson, S. Bertin, P. Hennig, and E. Lesellier, J. Chromatogr. A1440, 212–228 (2016).
- V. Desfontaine et al., J. Chromatogr. A1562, 96–107 (2018).
- B.W.J. Pirok, A.F.G. Gargano, and P.J. Schoenmakers, J. Sep. Sci.41, 68–98 (2018).
- D.R. Stoll and P.W. Carr, Anal. Chem.89, 519–531 (2017).
- B.W.J. Pirok and P. Schoenmakers, LCGC Europe31(5), 242–249 (2018).
- B.W.J. Pirok, D.R. Stoll, and P. Schoenmakers, Anal. Chem.91, 240–263 (2019).
- K. Ortmayr, S. Hann, and G. Koellensperger, Analyst140, 3465–3473 (2015).
- S. Wang, L. Zhou, Z. Wang, X. Shi, and G. Xu, Anal. Chim. Acta966, 34–40 (2017).
- T. Mairinger, T.J. Causon, and S. Hann, Curr. Opin. Chem. Biol.42, 9–15 (2018).
- V. D’Atri et al., J. Sep. Sci.41, 20–67 (2018).
- X. Zhang, K. Quinn, C. Cruickshank-Quinn, R. Reisdorph, and N. Reisdorph, Curr. Opin. Chem. Biol.42, 60–66 (2018).
- H.S. Jonasdottir et al., Anal. Chem.87, 5036–5040 (2015).
- R. Bonner and G. Hopfgartner, TrAC (2019) https://doi.org/10.1016/j.trac.2018.10.014.
- R. Wang, Y. Yin, and Z.J. Zhu, Anal. Bioanal. Chem.411, 4349–4357 (2019).
- R. Bonner and G. Hopfgartner, Bioanalysis8, 1735–1750 (2016).
- H. Zha, Y. Cai, Y. Yin, Z. Wang, K. Li, and Z.J. Zhu, Anal. Chem.90, 4062–4070 (2018).
- H. Tsugawa et al., Nat. Methods12, 523–526 (2015).
- E.A. Struys, Proc. Natl. Acad. Sci. U S A110, E4939 (2013).
- C.J. Venkatramani et al., Talanta148, 548–555 (2016).
- K. Sandra et al., Recent Developments in HPLC and UHPLC: LCGC Europe Supplement30(s6), 6–13 (2017).
Rob Haselberg is an assistant professor at the Vrije Universiteit Amsterdam, The Netherlands, where he focuses on the characterization of intact (bio)macromolecules, such as venom components, biopharmaceuticals, and industrial polymers. He recently expanded his work towards small molecule analysis in the forensic and bioanalytical context.
Bob Pirok obtained his M.Sc. degree in 2014, and prior to becoming a PhD student in 2015, worked at Shell. He received a string of international recognitions, including a Shimadzu Young Scientist Award at HPLC2015 Beijing, the Young Scientist Award Lecture during the SCM-8 meeting in Amsterdam in 2017, the Csaba Horváth Young Scientist Award at HPLC2017 Prague, the Journal of Chromatography A Young Scientist Award during the ISCC Conference in Riva de Garda in 2018, and the SCM Award at the SCM-9 meeting in Amsterdam in 2019. He has recently been appointed as assistant professor in the Analytical Chemistry group of the Van ‘t Hoff Institute for Molecular Sciences (HIMS).
Andrea Gargano is a tenure track assistant professor at the van’t Hoff Institute for Molecular Science, University of Amsterdam, The Netherlands. In his research, he works on the development of analytical chemistry technology for the characterization of macromolecules (keeping them intact). He received his M.Sc. in from the University of Pavia, Italy, and his Ph.D. from the University of Vienna, Austria, did his post-doctoral fellowship at the University of Amsterdam, The Netherlands, and has been guest research associate in several laboratories in Europe and the USA.
Isabelle Kohler studied pharmacy at the University of Geneva, Switzerland. She obtained her Ph.D. in pharmaceutical sciences in 2013 at the School of Pharmaceutical Sciences (University of Geneva), focusing on the use of capillary electrophoresis hyphenated to mass spectrometry in clinical and forensic toxicology. She carried out her postdoctoral fellowship at the Leiden University Medical Center, The Netherlands, in the Center for Proteomics and Metabolomics. Since July 2016, she has worked as an assistant professor in the group of Analytical Biosciences and Metabolomics at the Leiden Academic Center for Drug Research (LACDR).
Articles in this issue
almost 7 years ago
Vol 32 No 9 LCGC Europe September 2019 Regular Issue PDFalmost 7 years ago
Essential GC AccessoriesAdvertisement
Related Content
Advertisement




Advertisement
Advertisement
Trending on LCGC International
1
The Silent Crisis: Can The Demise of Chromatography Teaching in Universities Be Reversed?
2
HTC-19 Insights: Will At-Line Systems Outlast the Push for Full Automation?
3
GC-MS Reveals Regional Flavor and Aroma Shifts in Oranges
4
LC-MS Reveals Food-Linked Stress Proteins
5